Neurodegeneratív betegségek kémiai és biokémiai háttere
Penke Botond, az MTA lev. tagja
SZTE Orvosi Vegytani Intézet és MTA Fehérjekémiai Kutatócsoport, Szeged
penke@ovrisc.mdche.u-szeged.hu
1. Bevezetés
A neurodegeneratív betegségek nagy része fehérjeaggregátumok képződésével
kezdődik. Az aggregátumok részben a sejteken kívül helyezkednek el (ilyenek
pl. az Alzheimer-plakkok), részben viszont a sejten belül zárványokat alkotnak
(pl. a-synuclein a Lewy-testekben, hiperfoszforilezett
tau-fehérje neurofibrilláris kötegekben). A legismertebb neurodegeneratív
betegségeket és a jellemző fehérje aggregátumokat az 1. táblázat foglalja
össze.
Betegség
|
Fehérje- aggregátum
|
1. Alzheimer-kór
|
ß-amiloid (plakkok)
tau (neurofibrilláris kötegek)
|
2. Parkinson-kór
|
a-synuclein / ubiquitin
|
3. Lewy-testes demencia
|
a-synuclein
|
4. FTDP-17
|
tau (Pick-testek)
|
5. Huntington-kór
|
poli-glutamin / ubiquitin
|
6. Prion-betegség
(Creutzfeld-Jacob kór)
|
prion protein
|
7. Pick-betegség
|
tau (Pick-testek)
|
8. Amiotróf lateral sclerosis
|
ubiquitin
|
1. Táblázat A neurodegeneratív betegségek közös mechanizmusa:
fehérjeaggregátumok képződése
Egy és ugyanaz a fehérje aggregátum többféle betegségben is megjelenhet.
A tauopathiák nagy családja az Alzheimer kóron kívül a Pick-betegséget
valamint a frontotemporális demenciát és parkinsonizmust (FTDP-17) is magába
foglalja, az a-synuclein fibrillumok egyaránt
megjelennek a Parkinson-kórban és a Lewy-testes demenciában. Sokszor nem
könnyű a neurodegeneratív betegségeket egymástól elkülöníteni, éppen a
közös fehérje-aggregátumok miatt. Nagyon jellemző viszont az idegsejtek
károsodásának, pusztulásának a helye, így a legtöbb neurodegeneratív betegség
régió– sőt sejtspecifikus, legalábbis a betegség korai szakaszában. A kórképek
közös mechanizmusára utal az a tény, hogy a felsorolt fehérjék valamennyi
esetben konformációváltozást szenvednek (a-hélix
—> ß-szalag átalakulás) és ß-szerkezetű polipeptid láncokat tartalmazó
szálakká, fibrillumokká alakulnak át (Mager 2002). A fibrillumok, sőt a
kisebb, diffúzibilis aggregátumok is enzimrezisztensek és neurotoxikus
hatásúak. A szakirodalom megegyezik abban, hogy a felsorolt neurodegenerációs
betegségek végső oka bizonyos fehérjék nem megfelelő feltekeredése, gombolyodása
(misfolding), a hibás fehérje térszerkezetek kialakulása.
2. A neurodegeneratív betegségek genetikai háttere
Nem minden esetben tisztázott a neurodegeneratív betegségek genetikai háttere.
A 2. és 3. táblázat röviden összefoglalja a legfontosabb betegségek öröklődését
és az eddig ismert fontosabb mutáns géneket, genetikai faktorokat.
Betegség
|
Öröklődés, genetikai háttér
|
1. Alzheimer kór
|
Sporadikus/Autoszom. domináns
|
2. Parkinson kór
|
Sporadikus/Autoszom. recesszív
|
3. Lewy-testes demencia
|
Sporadikus/Autoszom. recesszív
|
4. FTDP-17
|
Autoszom. domináns
|
5. Huntington kór
|
Autoszom. domináns
|
6. Prion betegség
|
(részben öröklött)
|
7. Pick betegség
|
Sporadikus
|
8. Amiotróf lateral sclerosis
|
Sporadikus/Autoszom. domináns
|
2. Táblázat: A neurodegeneratív betegségek genetikai háttere
1.
Betegség
|
Mutáns Gének
|
1. Alzheimer kór
|
App, ps-1, ps-2, apoE e4,
tau, Gsk-3ß, ChAT, MTHFR
|
2. Parkinson kór
|
tau, N-acetil-transzferáz,
pank2, Mtdn 1-2
|
3. Huntington kór
|
(CAG ismétlődés)
|
4. Amiotróf lateral sclerosis
|
sod1, smn, tau
|
5. Down kór
|
tau, ChAT
|
3. Táblázat: A neurodegeneratív betegségek genetikai háttere
2.
A tisztán öröklött génhibán alapuló neurodegenerációs kórkép viszonylag
ritka, ilyen pl. a Huntigton-kór, amelynél egy poliglutamin-peptidlánc
(egy CAG triplett ismétlődése miatt fellépő rendellenesség) aggregációja
váltja ki az idegsejtek halálát. Ismerünk egy sorozat pontmutációt az a-synuclein
génben, amelyek kiváltják a familiáris Parkinson-kórt, illetve a Lewy-testes
demenciát. A neuronok mikrotubuláris rendszeréhez kapcsolódó tau-fehérjék
génjének pontmutációi abnormális hiperfoszforilációt idéznek elő, emiatt
a mikrotubuláris rendszer széthull, s ez nagymértékben hozzájárul a sejt
halálához. Az igen intenzív kutatómunka ellenére még sok esetben nem tisztázott
a neurodegeneratív betegségek genetikai háttere.
Az Alzheimer-kór a leggyakoribb demenciás kórkép, a memóriavesztéses
betegségeknek több mint 50%-áért felelős. A korai jelentkezésű (a 40-60.
életév között fellépő) familiáris Alzheimer-demencia (AD) előfordulása
5% alatt van. Az öröklődő betegségért néhány fehérje (pl. amiloid prekurzor
protein, presenilin-1 és 2, tau-fehérje) mutációi a felelősek, ezek a mutációk
jól ismertek, de igen ritkák. A mutációk az 1, 12, 14, 19 és 21. kromoszómában
lépnek fel (4. táblázat). Az Alzheimer-kór genetikai alapjáról Cacabelos
írt részletes összefoglalót (1999).
Kromoszóma
|
Gén
|
Jelentőség
|
Korkezdet
|
1
|
presenilin-2 (PS2)
1q 31-42
|
Korai familiáris AD,
autoszom. domináns
|
50-70
|
12
|
a2-makroglobulin
LRP1
|
késői AD
|
>75
|
14
|
presenilin-1(PS1)
14q 24.3, c-FOS,
14q 24.3-31
|
korai familiáris AD
autoszom.domináns
|
>40
|
19
|
APO-E e4
19q 12-23.3
|
késői familiáris AD
|
>55
|
21
|
amiloid prekurzor protein
21q 11.1-21.1
|
korai familiáris AD,
autoszom. domináns
|
>50
|
4. Táblázat: Az Alzheimer-kór genetikája: familiáris formák
A viszonylag ritka mutációk mellett az Alzheimer-kór gyakori és komoly
rizikófaktora az apo-E fehérje polimorfizmusa (Tariska, 2000). Ez a fehérje
299 aminosavból áll, főleg a lipidek transzportjáért és anyagcseréjéért
felelős. 3 különböző allélje van (e2, e3
és e4), amelyek csak 1-1 pontmutációban különböznek
egymástól, így egy vagy két aminosav különbséget találunk a polipeptid
láncban (5. táblázat). Az e2 allélnek megfelelő
fehérje (Cys 112, Cys 158) inkább védő hatású, viszont az e4
allélok (Arg 112, Arg 158) jelenléte 17-szeresére növeli az Alzheimer-kór
fellépésének gyakoriságát. Az eddigi vizsgálatok szerint az AD-esetek 15-20%-áért
az apo-E e4 allél jelenléte a felelős.
|
112. hely
|
158. hely
|
APOE allél
|
Triplet
|
Aminosav
|
Triplet
|
Aminosav
|
e2
|
TGC
|
Cys
|
TGC
|
Cys
|
e3
|
TGC
|
Cys
|
CGC
|
Arg
|
e4
|
CGC
|
Arg
|
CGC
|
Arg
|
5. Táblázat: Az apo-E gén polimorfizmusa mint az Alzheimer-kór
kiváltó oka
Ha valamennyi ma ismert genetikai faktort összeadunk, azt találjuk,
hogy az Alzheimer demenciák 20-25%-át genetikai tényezők váltják ki. A
késői kezdésű (65. életév után jelentkező) AD-k legnagyobb részénél nem
ismerjük a kiváló okokat. A betegség többtényezős, multifaktoriális eredetűnek
látszik. A neurotoxikus b-amiloidok szintéziséért,
ill. lebontásáért felelős proteázok egyensúlyának zavara, a szabad gyökök
megkötéséért felelős enzimek alulműködése, az idegsejtekben folyó ATP-termelés
csökkenése, a vér-agy gát permeábilitás megnövekedése egyaránt hozzájárulhatnak
a betegség kialakulásához. Magyarországon az Alzheimer-kór különösen gyakran
társul vaszkuláris eredetű demenciával; az agyi erek szklerózisa az Alzheimer-kór
egyik rizikófaktora. Bizonyos vaszkuláris faktorok változása (pl. magas
vérnyomás, az agyi kapillárisok állapota, stb) fontos kóroki tényező lehet.
A legújabb kutatások kiderítették, hogy az Alzheimer-kór kialakulása igen
hosszú folyamat (15-20 év), emiatt igen valószínű, hogy a középkorú populáció
(40-55. év) kezeletlen magas vérnyomásos betegeinél nagymértékben megemelkedik
az AD kockázata. Komoly kockázati tényező az életkor előrehaladása, az
elszenvedett koponyatraumák és a tartós oxigénhiány.
3. A leggyakoribb neurodegenerációs betegség, az Alzheimer-kór
morfopatológiája, biokémiája és kialakulásának mechanizmusa
Az Alzheimer-demenciát Alois Alzheimer már 1907-ben leírta és a következő
jellemzőit sorolta fel: az agyszövet nagyfokú atrófiája; amiloid plakkok
kialakulása bizonyos agyterületeken, ill. neurofibrilláris kötegek megjelenése.
A betegség elsősorban a szinapszisok és a kolinerg neuronok pusztulásával
jár (Selkoe 1991). Feltűnő a neuronok mitokondriumainak sérülése, elfajulása.
Ez komoly ATP-hiányt okoz, többen ezt tartják az idegsejt pusztulás végső
okának. Az igazi okokat azonban jóval korábbi lépéseknél kell keresni.
Nagyon sokan vizsgálták a (csak mikroszkóppal látható) amiloid plakkok
szerkezetét és összetételét. A betegség előrehaladásával a plakkok is változnak:
a kezdetben diffúz plakk több lépésben szenilis plakká alakul, közepén
„keményítőszerűen” festődő maggal (innen jön az amiloid név). A mag kissé
szivacsos állományú és főleg b-amiloid peptideket,
tau-fehérjét, lipofuscint és más anyagokat tartalmaz. Bizonyos festékek
(pl. Kongóvörös, tioflavin) specifikusan kötődnek a b-amiloidokhoz,
ennek az a magyarázata, hogy a plakkokban a polipeptidek ún. b-redőzött
réteg vagy b-szalag szerkezetet vesznek fel.
Ezek egymáshoz kapcsolódnak, aggregálódnak és hosszú fibrillumokat alakítanak
ki. Az amiloid aggregátumok neurotoxikus hatásúak: a plakk közelében húzódó,
a plakk magjával érintkező axonok degenerációját indítják el. (A sejttest
sokkal kevésbé érzékeny a b-amiloid aggregátumokra).
A betegség előrehaladásával, súlyosodásával szinte egyenes arányban nő
az elhalt idegsejtekből képződő neurofibrilláris kötegek mennyisége, ezeket
főleg a már említett „abnormálisan” foszforilezett (túl sok foszfátészter-csoportot
tartalmazó) tau-fehérjék alkotják. Az amiloid plakkok száma nem áll mindig
arányban a betegség súlyosságával.
Az Alzheimer-kór az öregkor betegsége, de ritka esetben fiatalabb korban
is jelentkezik. Évtizedes vita után el kell fogadnunk, hogy a betegséget
a b-amiloid peptidek túltermelődése, aggregációja
váltja ki, ez az indító lépés. (Ez nem áll ellentétben azzal a ténnyel,
hogy bizonyos tau-fehérje mutációk b-amiloid
képződés nélkül is neurodegenerációhoz vezetnek, a tau-fehérjék hiperfoszforilezése
révén. Ezt a betegséget a plakkok hiánya miatt nem tekinthetjük Alzheimer-kórnak).
A fiatalabb korban, a 40-65. életév között jelentkező Alzheimer-kórt főleg
az amiloid prekurzor protein (APP) és a presenilinek mutációi idézik elő
(4. táblázat): ezek hatására az APP-ből nagy mennyiségű, igen könnyen aggregálódó
neurotoxikus peptid képződik. Az APP egy sejtadhéziós fehérje, a szinaptikus
membránokban van jelen nagy koncentrációban, pontos biológiai szerepét
nem ismerjük. Különböző molekuláris formái vannak: a neuronokban a 695
aminosavas, a glia sejtekben a 751 ill. 770 aminosavas forma fordul elő
(Tanzi, 1988). A központi idegrendszert ért traumák hatására az APP nagy
mennyiségben szabadul fel. Állandóan ismétlődő agyi traumák ill. hipoxia
hatására sok APP termelődik, ez nagyfokú b-amiloid
képződést okoz. Ezzel magyarázzák a boxolók dementia pugilisticaját, de
a gyakori hipoxiás állapotba kerülő sportolók (hegymászók ill. búvárok)
korai demenciáját is.
Az idegsejtek elhalásának pontos mechanizmusát még nem sikerült teljesen
igazolni Alzheimer-kór esetén. Sejtszinten, molekuláris szinten a következő
lépésekben képzeljük el a betegség kialakulását az amiloid-kaszkád hipotézis
alapján (Hardy, 1992).
1. Kisebb-nagyobb agyi traumák, hipoxia, esetleg genetikai
faktorok APP túltermelődéshez vezetnek, a prekurzorból a normálisnál nagyobb
mennyiségű b-amiloid peptid képződik. Idősebb
korban ezt elősegíti a lebontó proteázok csökkent működése is.
2. A sejtek felszínén, az extracelluláris térben a b-amiloid
peptidek neurotoxikus aggregátumokat képeznek (Lambert, 1998). Ezek különböző
nagyságúak, a kisméretű, diffúzibilis aggregátumoktól a hosszú szálakig
sokféle forma előfordulhat, de valamennyi forma toxikus. (Maguk a monomer
b-amiloidok
nem toxikusak.)
3. A b-amiloid aggregátumok megkötődnek az
idegsejtek membránfehérjéin. Valószínű, hogy a b-amiloid
aggregátumnak klasszikus értelemben nincs receptora, hanem többféle fehérjén
meg tud kötődni. (Van olyan vélemény, hogy a b-amiloid
aggregátum számára „minden membránfehérje kötőhely”, de ezt a kísérletek
nem igazolják). A fehérjék egy része G-proteinnel kapcsolt receptor.
4. A b-amiloid-membránfehérje kötődés hatására
Ca2+ ionok áramlanak be a sejtbe. Igazolt, hogy az amiloid peptidek
megkötődnek az NMDA-receptoron, az integrin-receptorok bizonyos típusain,
az APP-n, a RAGE-receptoron, stb. Mivel az amiloid aggregátum enzimrezisztens
és tartósan ott marad a membránon, a Ca2+-beáramlás állandósul.
5. A Ca2+-jel aktiválja a protein kinázokat (pl. Cdk5, GSK3
b)
és megkezdődik a mikrotubuláris-rendszert alkotó tau-fehérjék abnormális
helyen történő foszforilezése (hiperfoszforileződés). Eltolódik a foszforiláz-foszfatáz
enzimegyensúly, az abnormális tau-fehérjék nem képesek a mikrotubulusok
szervezésére, a szerkezet összeomlik. A hiperfoszforilezett tau-fehérjék
lassan neurofibrilláris kötegekké aggregálódnak.
6. A megemelkedett Ca2+-szint egyedül is elegendő a mitokondriumok
károsításához. A kettős membrán felszakad, a sejtlégzés és az ATP képződés
leáll, nagy mennyiségű szabad gyök képződik. A mitokondiumból kiszabaduló
faktorok (apoptózis indukáló faktor, citokróm-c) beindítják a neuron elhalását.
7. Az axonok is központi szerepet játszanak a neurodegenerációban. A
mikrotubuláris rendszer összeomlásával megszűnik az axonális transzport.
A neuron lassan elveszíti dendritjeit és axonját, legömbölyödik (dezarborizáció,
vezikularizáció) és lassan elhal.
4. Az Alzheimer-kór kezelése, megelőzések lehetőségei
Az elhalt neuronokat már nem lehet visszahozni, viszont a patomechanizmus
ismeretében ma már nem reménytelen a betegség kezelése és a racionális
gyógyszertervezés.
A kolinerg rendszer részleges kiesését, az acetilkolin-szint csökkenését
kolinészteráz-gátlókkal próbálják kivédeni, ez természetesen csak tüneti
kezelést jelent. Újabb kezelési lehetőség a Memantine (dimetil-Amantadin)
alkalmazása. Ez a gyógyszer bekötődik az NMDA-receptor ioncsatornájába
és megakadályozza a Ca2+-beáramlást. A szakirodalom igen jó
eredményekről számol be: ha az idegsejtek még nem haltak el, csak „fojtogatja”
őket az amiloid aggregátum, a Ca2+-beáramlás megakadályozásával
ezek a sejtek „felélednek”, visszanyerik működőképességüket és Memantine
hatására a betegek állapota javul.
A szakirodalom részletesen beszámol a reaktív szabad gyököket „eltakarító”
anyagok (C-vitamin, E-vitamin, flavonoidok) kedvező hatásáról is. Ezek
mellett a szteroid-hormonok szerepével, hatásmechanizmusával is sokan foglalkoznak.
Az Alzheimer-kutatás legújabb iránya abból indul ki, hogy a ß-amiloid
peptidek központi szerepet töltenek be a betegség kialakulásában, ezek
keletkezését, aggregációját ill. sejtmembránhoz való kapcsolódását kell
megakadályozni.
A ß-amiloid peptidek képződésének gátlása
A prekurzor fehérje lebontásában 3 enzim játszik kulcsszerepet: az a-,
b-
és g-szekretáz. Ha az a-szekretáz
hasít, az APP-ből vízoldható, nem toxikus peptidek sokasága képződik. Mindig
van lehetőség viszont az alternatív hasításra: a b-szekretáz,
majd g-szekretáz működése olyan, 40-42 aminosavból
álló peptideket hasít ki az APP-ből, amelyek igen könnyen aggregálódnak
s emiatt neurotoxikusak (b-amiloidok). A b-amiloid
monomerek kis mennyiségben mindig képződnek és neuromodulátor hatásúak:
csökkentik a kolinerg-receptorok ingerelhetőségét. Az alternatív hasítás
csak akkor veszélyes, ha nagy mennyiségű
b-amiloidot
termel és ez aggregálódik. Megfelelő enzimgátlókkal a b-amiloid
képzés csökkenthető és (elvileg) a kór előrehaladása megakadályozható.
A b-szekretáz egy aszpartil-proteáz (Vassar,
1999), Röntgen-diffrakciós szerkezete ismert. Számos laboratóriumban (így
nálunk is) folyik a specifikus b-szekretáz inhibitorok
számítógépes tervezése és szintézise. Úgy tűnik, a b-szekretáz
gátlása nem okoz különös mellékhatásokat kísérleti állatokon.
A g-szekretáz szintén aszpartil-proteáz,
egy bonyolult membránfehérje-komplex. Az az érdekessége, hogy a polipeptidláncot
éppen a membrán belsejében középen hasítja, az APP-transzmembrán régiójában.
A g-szekretáz Röntgen-diffrakciós szerkezete
nem ismert, ennek ellenére számos inhibitorát ismerjük a szakirodalomból.
A sejtmembrán lipid összetétele nagymértékben befolyásolja a b-
és g-szekretázok aktivitását. Nagy mennyiségű
koleszterin jelenléte a membránban növeli a b-
és g-szekretáz aktivitását, így a koleszterin
bioszintézist gátló gyógyszerek (Lovastatin, Mevastatin, stb.) jó hatással
lehetnek Alzheimer-kórban. Ugyanakkor a többszörösen telítetlen w-3
zsírsavak (dokoza-hexaénsav, C22:6, DHA és eikozapentaénsav, C20:5, EPA)
jelenléte a membránban csökkenti a b- és g-szekretáz
aktivitást és a keletkező b-amiloidok mennyiségét.
Intenzív kutatómunka folyik olyan diéta kidolgozására, amelyik többszörösen
telítetlen zsírsavak bevitelével akadályozza meg az Alzheimer-kór kialakulását,
illetve lassítja le a betegség előrehaladását.
A b-amiloidok aggregációja
és toxicitása
A b-amiloidok aggregációja az Alzheimer-kór
fontos rizikófaktora. Megvizsgáltuk, hogyan függ össze az amiloid peptidek
szerkezete, aggregációs képessége és neurotoxicitása. Az aggregációt FT-IR
spektroszkópiával követtük nyomon, a neurotoxicitást differenciált SH-SY5Y
neuroblasztóma tenyészeten MTT-teszttel mértük. (A teszt a sejtek életképességét,
redukciós potenciálját méri, egy tetrazolfesték formazánná történő redukciós
átalakulásával). Megmértük a különböző lánchosszúságú b-amiloid
peptidek, valamint a csupán D-aminosavat tartalmazó peptidek, illetve a
fordított (reverz) aminosav-sorrendű analógok aggregációs készségét és
toxicitását. Az eredményeket az 1. és a 2. ábra mutatja.
Szekvencia
|
Aggregáció
|
Toxicitás
|
1. Ab 1-40
|
+++
|
+++
|
2. Ab 1-42
|
++++
|
++++
|
3. Ab 25-35
|
+++
|
+++
|
4. Ab 31-35
|
+++
|
+++
|
5. csupa D-Ab 1-40
|
+++
|
+++
|
6. reverz Ab (42-1)
|
—
|
—
|
7. reverz Ab (35-25)
|
—
|
—
|
1. ábra: Amiloid peptidek aggregációja és toxicitása.
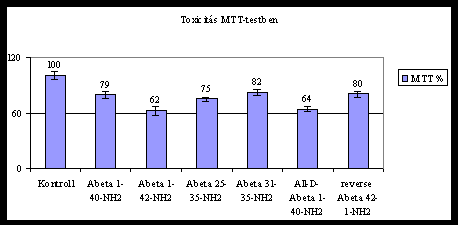
2. ábra: Ab-peptidek toxicitása
MTT-teszten
Az Ab-peptidek toxicitása jó korrelációban
van az aggregációjukkal. Néhány kis Ab-fragmens
is toxikus (25-35, 31-35). A csupán D-aminosavakból felépülő Ab
1-40 is toxikus, mivel gyorsan aggregálódik. Ezzel szemben a fordított
sorrendű Ab 42-1 nem képez aggregátumokat és
alacsony toxicitást mutat.
Toxikus amiloid-aggregátumok képződésének megakadályozása
A b-amiloid polipeptid láncához különböző típusú
vegyületek kapcsolódhatnak ionos kötéssel ill. másodlagos kötésekkel. Az
ilyen vegyületek megakadályozzák a peptidlánc aggregációját és elvileg
alkalmasak lehetnek az Alzheimer-kór kezelésére. Ezeket az anyagokat összefoglaló
néven b-szerkezetrombolóknak (b-sheet-breaker)
nevezzük. Legismertebb ezek közül az azofesték jellegű Kongóvörös.
Tjernberg (1996), majd később Soto (1996) kutatócsoportja ismerte fel,
hogy a b-amiloid 16-20, ill. 17-21. pentapeptid
részlete megakadályozza az aggregációt. Számos módosított b-szerkezetromboló
peptidet állítottak elő, ezek közül a legismertebb a Leu-Pro-Phe-Phe-Asp
pentapeptid.
Kutatócsoportunkban számítógépes molekulatervezéssel, a b-amiloid
peptidlánc felszínére történő illesztéssel (dokkolás, AUTODOCK-program)
kerestünk olyan peptideket, amelyek viszonylag erős kötéssel illeszkednek
az amiloidhoz és megakadályozzák az aggregációt (3. ábra). Ezeket szintetikusan
is előállítottuk, majd in vivo és in vitro tesztekben megvizsgáltuk a szintetikus
peptidek neuroprotektív hatását.
A következő vegyületek bizonyultak legjobbnak:
Leu-Pro-Tyr-Phe-Asp
Arg-Val-Val-Ile-Ala
Ezek a vegyületek önmagukban is potenciális gyógyszerek. A vér-agy gáton
áthaladó, enzimrezisztens analógok és peptidomimetikumok tervezése és szintézise
szintén megkezdődött. A dokkolás egyik lehetőségét a 3. ábra mutatja be.
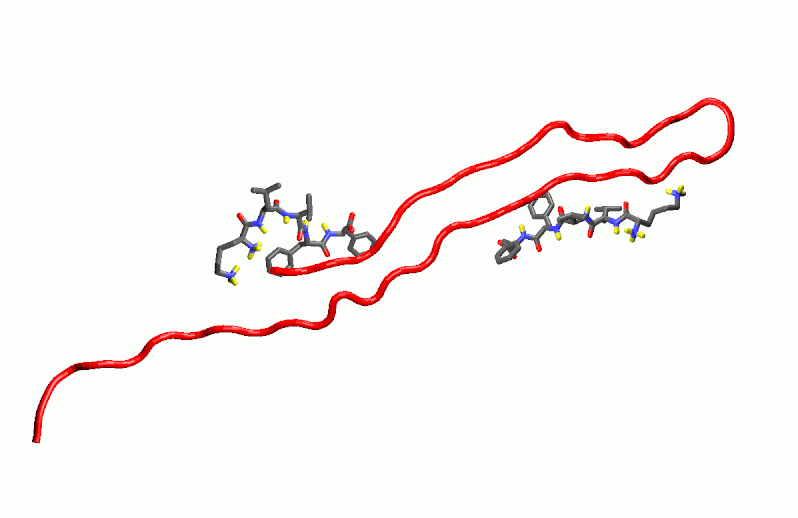
3. ábra: A Lys-Leu-Val-Phe-Ale-amid dokkolása az Aß 1-42 peptidre
Az aggregált amiloid és a membránfehérjék közötti kötődést
gátló neuroprotektív anyagok: funkcionális antagonisták
Még évekkel ezelőtt azt találtuk, hogy a b-amiloid
egy rövid fragmense, az Ile-Ile-Gly-Leu tetrapeptid-amid és származékai
megakadályozzák a b-amiloid peptidek neurotoxikus
hatásának kifejlődését (Laskay, 1997).
Mivel ezek a peptidek nem b-szerkezetrombolók,
feltételeztük, oly módon hatnak, hogy megakadályozzák az aggregátumok kötődését
a membránfehérjéken, így a Ca2+-ionok beáramlását és az azt
követő apoptózishoz vezető biokémiai folyamatokat. A vizsgálatok igazolták
ezt az elképzelést, így erre a vegyületcsoportra egy új nevet kellett bevezetnünk:
ezek az ún. ASBIM (Amyloid Surface Binding Molecule) vegyületek. In vitro
és in vivo tesztekben a következő peptidek bizonyultak legjobbnak:
propionil-Ile-Ile-Gly-Leu-amid
Arg-Ile-Ile-Gly-Leu-amid
Phe-Arg-His-Asp-Ser-amid
Ezek a vegyületek a b-amiloidok funkcionális
antagonistáinak tekinthetők. Valamennyi peptid tartalmaz b-amiloid
szekvencia részletet. Az utolsó két vegyület ún. RGD analóg, tehát az integrin-fehérjékhez
kötődve is kifejtheti hatását. Ezek a vegyületek vezérvegyületként szolgálnak
olyan enzimrezisztens, a vér-agy gáton áthaladó peptidomimetikumok tervezéséhez
és szintéziséhez, amelyek az Alzheimer-kór igazi gyógyszerei lehetnek.
Összefoglalva: az Alzheimer-kór patomechanizmusának ismeretében új utak
nyíltak a gyógyszertervezés előtt: enzimgátlók, b-szerkezetrombolók
és a membránfehérje – aggregált amiloid kölcsönhatást gátló vegyületek
lehetnek a jövő potenciális gyógyszerei. Valószínű, hogy a fehérjeaggregátumok
keletkezését és toxicitását megakadályozó vegyületek alkalmazásának alapelve
kiterjeszthető a többi neurodegeneratív betegség megelőzésére is.
Irodalom
Cacabelos, R.: Association of genetic risk factors in Alzheimer’s disease.
In: Iqbal, K. (ed) Alzheimer’s Disease and Related Disorders Wiley,
Chichester. (1999) 93. p.
Hardy, J. A, Higgins, G.A.: Alzheimer’s disease: the amyloid cascade
hypothesis. Science 256 (1992) 184. p.
Lambert, M. P., Finch, C. E., Krafft, G. A., Klein, W. L.: Diffusible,
nonfibrillar ligand derived from Aß 1-42 are potent central nervous system
neurotoxins. Proc. Nat. Acad. Sci. USA 95 (1998) 6448. p.
Laskay, G., Zarándi, M., Varga, J., Jost, K., Fónagy, A., Torday, C.,
Latzkovits, L., Penke, B.: A putative tetrapeptide antagonist prevents
ß-amyloid induced long-term elevation of [Ca2+]i.
Biochem.
Biophys. Res. Commun. 235 (1997) 479. p.
Mager, P.P., Penke, B., Walter, R., Harkány, T. and Härtig, W.: Pathological
Peptide Folding in Alzheimer’s Disease and Other Conformational Disorders.
Current Medicinal Chemistry 9 (2002) 1763. p.
Selkoe D. J.: The molecular pathology of Alzheimer’s disease. Neuron
6
(1991)
487. p.
Soto, C., Kindy, M. S., Baumann, M., Frangione, B.: Inhibition of Alzheimer’s
amyloidosis by peptides that prevent ß-sheet conformation. Biochem.
Biophys. Res. Comm. 226 (1996) 672. p.
Tanzi R. E., McClatchey A. I., Lamperti E. D., Villa-Komaroff L., Gusella
J. F., Neve R. L.: Protease inhibitor domain encoded by an amyloid protein
precursor mRNA associated with Alzheimer’s disease. Nature 331
(1988)
528. p.
Tariska, P.: Alzheimer kór. Golden Book, Budapest (2000)
Tjernberg, L. O., Naslund, J., Lindquist, F., Johansson, J., Karlstrom,
A. L., Thyberg, J., Terenius, L., Nordstedt, C.: Arrest of ß-amyloid fibril
formation by a pentapeptide ligand. J. Biol. Chem. 271
(1996)
8545. p.
Vassar, R.: ß-secretase cleavage of Alzheimer’s amyloid precursor protein
by the transmembrane aspartic protease BACE. Science 285 (1999)
735. p.
<