Homogénkatalitikus hidroalkoxikarbonilezés palládium
tartalmú rendszerekkel
Veszprémi Egyetem, Szerves Kémia Tanszék
Veszprém
2000
I. Elõzmények, célkitûzés
A szén-szén kötések kialakításával járó reakciók a szintetikus szerves kémia alapvetõ törekvéseinek részei. Az olefinek hidrokarboxilezése, illetve hidroalkoxikarbonilezése fél évszázada ismert, az ipari gyakorlatban is alkalmazott eljárások, melynek során olefinekbõl savak vagy észterek állíthatók elõ. A termékek közé tartoznak olyan alapvetõ vegyipari anyagok mint a propionsav, adipinsav (BASF). Napjainkban a szerves molekulák szénmonoxiddal történõ funkcionalizálása dinamikusan fejlõdõ terület, melynek további kiszélesedése várható az új katalizátorok kifejlesztésének, a mechanizmus mélyebb megértésének nyomán.
Királis információt hordozó katalizátor általában palládium-foszfin komplex jelenlétében lehetõség nyílik optikailag aktív észterek (vagy szerves savak) elõállítására prokirális olefinekbõl. Noha a kutatók az aszimmetrikus hidrogénezéshez képest valamivel kevesebb figyelmet fordítottak a hidroészterezésre, e téren is született néhány ígéretes eredmény.
Ilyen megfontolás adta az ötletet biológiai jelentõségû szubsztrátumok alkoxikarbonilezési reakcióinak tanulmányozásához is. Mint ilyenek, a növényvilágban elterjedt terpének elsõsorban az illatszer-, illetve a kozmetikai ipar szempontjából jelentõsek, de herbicid tulajdonságaik is ismeretesek. Másfelõl a prokirális anyagokból elõállítható optikailag aktív észterek potenciálisan biológiailag aktívak, vagy ilyen vegyületek építõelemei lehetnek, ennek ellenére az irodalomban nem találtam a terpének diasztereoszelektív hidroalkoxikarbonilezésére irányuló próbálkozásokat. Ennek szellemében, az általam végzett munka a vegyületcsalád néhány képviselõjének aszimmetrikus hidroészterezését célozza meg.
Eredményeimhez jó összehasonlítási alapnak kínálkozott a kézenfekvõ biológiai aktivitással rendelkezõ vinil-ösztron, mely a vinil-aromás váz modelljeként szolgált.
A modern kémiában kulcsfontosságú szerepet játszik az egyes reakciók mechanizmusának minél részletesebb megismerése és megértése, a reakció során regisztrált eredmények (konverzió, kemo-, regio- és enantioszelektivitás) interpretációja. Ennek megfelelõen munkám második részében célomul a tanulmányozott reakció izotóppal (jelen esetben 2H) néhány egyszerû modellvegyületen történõ nyomonkövetését tûztem ki. A módszer létjogosultságát a spektroszkópiai technikák (GC-MS, NMR) megfelelõ kombinációja teremti meg. Ezek segítségével kívántam nyomon követni a termékekben fellelhetõ izotópdúsulás helyét és mennyiségét, melyek fényt derítenek a hozzájuk vezetõ lehetséges reakcióutakra, illetve ezek közbensõ termékeire.
Dolgozatomban az említett modellvegyületek szelektív
átalakítását és a reakció mechanizmusának
tanulmányozását céloztam meg. Ez utóbbi
jobb megértése segíthet a kísérleti
eredmények értékelésében és új
impulzust adhat a további kísérleti munkához.
II. Alkalmazott kísérleti módszerek
Az általam végzett kísérletek mindegyike víz- és oxigénmentesített oldószereket, kiindulási anyagokat igényelt. Ezek elõállítását ismert módszerekkel valósítottam meg, a bemérések során minden esetben Schlenk-technikát alkalmaztam.
A szerkezeti vizsgálatok javarészét (1H, 2H, 13C) NMR spektroszkópiával, illetve tömegspektrometriásan végeztem. A terpénekbõl képzett észterek diasztereomerjeit HPLC technikával választottuk el.
A hidroalkoxikarbonilezési kísérletek során
keletkezõ termékelegy összetételét gázkromatográfiás
módszerrel határoztam meg.
III. Új tudományos eredmények
1. Megállapítottam, hogy az általam vizsgált monoterpének (limonén, karvon, dihidrokarvon, pulegon) jó kemo- és regioszelektivitással hidroalkoxikarbonilezhetõk az alkalmazott Pd-foszfin katalizátor rendszerek jelenlétében. A reakció során az új aszimmetrikus szénatomot tartalmazó egyenes láncú termék keletkezik. E szubsztrátumok belsõ aszimmetrikus indukciója az egyszerû vinil-aromásokhoz képest igen szerény diasztereoszelektivitáshoz vezet, melyet királis katalizátorral sem sikerült jelentõsen befolyásolni.
2. Módszert dolgoztam ki a keletkezett diasztereomerek HPLC technikával történõ elválasztására.
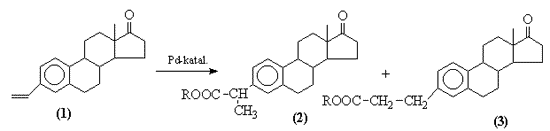
3. Modellvegyületként vinil-ösztront alkalmazva vizsgáltam a hidroalkoxikarbonilezés kemo-, regio- és diasztereoszelektivitását néhány akirális és királis palládium-katalizátor jelenlétében. Az alkalmazott rendszerekben általában az új sztereogén centrummal rendelkezõ elágazó láncú észter dominál. A keletkezett új vegyületeket analitikai módszerekkel (MS, NMR) azonosítottam.
A szubsztrátum szerkezetében jelen levõ aromás szerkezeti elem kedvezõ hatásának köszönhetõen kiemelkedõ diasztereoszelektivitást (70%) értem el. Akárcsak a terpének esetében, vinil-ösztron hidroalkoxikarbonilezési reakciójában is elsõsorban a kiindulási molekula belsõ indukciója határozza meg az optikai hozamot, a királis katalizátor alárendelt szerepet kap.
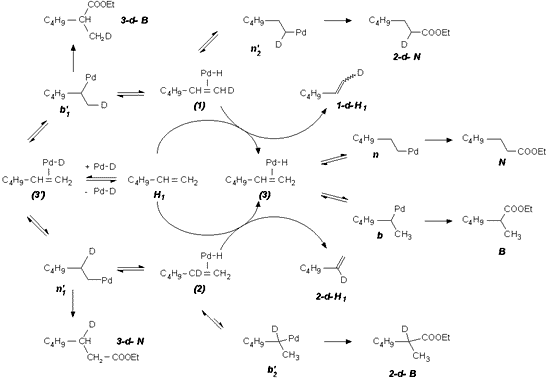
4. Sztirolt Pd(PPh3)2Cl2/SnCl2 rendszerben deuterioalkoxikarbonilezve módszert (MS, NMR technikák) dolgoztam ki a keletkezett deuterált speciesek gyors és egyértelmû kimutatására és az izotópdúsulás meghatározására.
5. A fenti eredmények interpretációja során az adott reakciókörülmények között a "hidrido" mechanizmus mûködését valószínûsítettem.
(a) Megállapítottam, hogy az izotóp beépülése a karbonilezési lépést megelõzõ reverzibilis olefin inzerció/ß-hidrid-elimináció eredménye.
(b) Az átalakulatlan szubsztrátum deutérium tartalma a reakcióutak reverzibilitására és az alkil-palládium s-komplexek disszociációjára utal.
(c) A keletkezett összes deuterált species keletkezése megmagyarázható a feltételezett reakcióutakkal.
6. Sztirol Pd(PPh3)2Cl2 és Pd(PPh3)2Cl2/SnCl2 prekurzorokkal végzett deuterioalkoxikarbonilezésénél a nagymértékû regioszelektivitásbeli különbséget a fém-alkil közbensõ termékek eltérõ viselkedése okozza.
(a) Pd(PPh3)2Cl2 jelenlétében a palládium-komplex anti-Markovnyikov addícióját a konkurrens elektronikus faktorok (a fenilgyûrû elektronvonzó hatása következtében fellépõ polarizáció, a fenilgyûrû és a palládium p-benzil kölcsönhatása) gátolják, ennek következtében az elágazó láncú termék dominál. Az egyenes láncú Pd-alkil komplexekhez vezetõ reakcióutak irreverzibilisek.
(b) Az SnCl2 jelenlétében létrejött SnCl3- ligandum sztérikusan gátolt katalizátort eredményez, mely túlkompenzálja a kedvezõtlen elektronikus tényezõket. Ebben a rendszerben a fém-alkil intermedierek átalakulásai minden esetben reverzibilisek.
7. Az a -metil-sztirol deuterioalkoxikarbonilezése során detektált deutérium beépüléssel bizonyítható, hogy a reakcióban tercier Pd-s-komplex képzõdik.
8. A fenti szubsztrátum esetében a sztirolétól jelentõsen eltérõ regioszelektivitás oka az eredetileg kedvezményezett tercier Pd-alkil gyors ß-eliminációja. Az átizomerizálás következtében az egyenes láncú észter keletkezik túlsúlyban.
9. Megfelelõen megválasztott módszer (kémiai ionizáció) segítségével lehetõség nyílt 1-hexén deuterioalkoxikarbonilezési termékeinek tömegspektrometriás meghatározására. Az MS és NMR technikák alkalmazása lehetõvé tette 1-hexén deuterioalkoxikarbonilezési reakciójának nyomonkövetését.
10. Megállapítást nyert, hogy alkil-olefin esetében
Pd(PPh3)2Cl2 jelenlétében
a terminális fém-alkil intermedier nem vesz részt
átrendezõdési folyamatokban, ami magyarázatot
ad a hõmérséklet emelésével tapasztalt
regioszelektivitásbeli változásra, vagyis az elágazó
láncú termék arányának csökkenésére.
Ha a rendszerben SnCl2 van jelen, a lineáris s-komplex
jelentõs izomerizációja megy végbe már
alacsony hõmérsékleten is, ami az egyenes láncú
észter mennyiségének csökkenését
okozza a körülmények erélyesebbé válásával.
A jelenségek az alábbi feltételezett reakcióutakkal
magyarázhatóak.
IV. Tudományos eredmények jelentõsége
Noha doktori munkám tárgya az alapkutatás területéhez tartozik, az elõállított új termékek egyrészt lehetõséget kínálnak a váz továbbépítésére, másrészt mint potenciális gyógyszeralapanyagok vehetõk figyelembe. A palládium-katalizált hidroalkoxikarbonilezés mechanizmusának ismerete elõnyt jelenthet szelektív katalizátorrendszerek kifejlesztésénél, a reakció paramétereinek megtervezésénél. A különbözõ szerkezetû modellvegyületekbõl a deuterizálás módszerével nyert intermedierek, az ezek segítségével felépített reakció-utak a reakció mechanizmusának néhány új elemét bizonyították, melyek segítik a különbözõ katalizátorrendszerekkel elért eltérõ regioszelektivitások jobb megértését.
Ezúton fejezem ki köszönetemet témavezetõmnek, Dr. Heil Bálintnak munkám irányításáért, áldozatkész segítségéért.
Homogeneously catalyzed hydroalkoxycarbonylation in presence of palladium-containing systems
Ph.D. thesis
University of Veszprém
Department of Organic Chemistry
Veszprém
2000
I. Preliminaries, aims of the work
The carbon-carbon bond forming reactions are of basic interest in synthetic organic chemistry. The hydrocarboxylation and hydroalkoxycarbonylation of olefins have been known for decades as industrially applied processes (Monsanto, BASF) for the production of acids or esters from olefins. As a dinamically expanding field, the functionalization of organic molecules with carbon monoxide is continuously stimulated by the development of new, highly selective catalysts and a better understanding of the mechanism of the reactions.
In presence of a chiral catalyst generally a palladium-phosphine complex in this reactions it is possible to prepare optically active esters (or acids) from prochiral olefins. Although hydroesterification has not been studied as intensively as asymmetric hydrogenation, some promising results have been reported.
Similarly, we were interested in applying selective hydroalkoxycarbonylation to transform some biologically important substrates. As some of these, terpenes are used mainly in perfumery and cosmetical industry, but their herbicidal properties have also been proved. Although their optically active ester derivatives are potentially pharmacologically active, diastereoselective hydroalkoxycarbonylation of terpenes hasn't been reported yet. Arising from this fact, part of this work deals with asymmetric hydroesterification of some terpenes.
These results have been compared with the data obtained in transformation of vinyl-estrone, a model of a vinyl-aromatic skeleton of obvious biological importance.
The knowledge of the reaction mechanism is crucial in modern chemistry, as well as interpretation of the results obtained (conversion, chemo-, regio- and enantioselectivity). As a consequence, in the second part of the dissertation efforts have been made to determine the routes governing the reaction. As a method, deuterization has been applied using some simple molecules as models. The accesibility of this method has been realised through the combination of MS and NMR analysis which enabled a rapid and exact determination of the amount and the distribution of deuterium atoms in the compounds present. Thus, the isotope incorporation throws light on the possible reaction routes and the intermediates leading to the products detected.
In summary: the work aims the selective transformation of the model
compounds used and the investigation of the reaction mechanism. A better
understanding of this latter may be helpful in evaluation of the experimental
data and provide new impulse to further studies.
II. Experimental methods applied
All the experiments required the use of water and oxigen free solvents and starting materials. The preparation of these has been executed according to known methods. Carrying out the reactions, Schlenk technique has been applied in each case.
Most of the analyses have been based on (1H, 2H, 13C) NMR spectroscopy and mass spectrometry. The epimers resulted in transformation of terpenes have been separated by HPLC.
The reaction mixtures resulted in hydroalkoxycarbonylation have been
determined by gas chromatography.
III. New scientific results
1. It has been proved that hydroalkoxycarbonylation of the monoterpenes investigated (limonene, carvone, dihydrocarvone, pulegone) can be carried out with good chemo- and regioselectivities in presence of the Pd-phosphine catalyst systems applied. The reaction results in the formation of the linear product containing a new asymmetric carbon atom. The diastereoselectivity of these reactions is, however, very low, differing significantly from that observed at aromatic molecules. This feature origins from the unfavourable internal asymmetric induction of the substrates which can be only slightly influenced by the choice of the chiral catalyst.
2. HPLC method has been elaborated for separation of the epimers resulted.
3. Using vinyl-estrone as a model, chemo-, regio- and diastereoselectivity of hydroalkoxycarbonylation has been investigated in presence of some achiral and chiral palladium catalysts. In the systems applied the branched ester possessing a new stereogenic center dominates. The new products resulted have been identified by analytical methods (MS, NMR).
Excellent diastereoselectivity (70%) has been reached in hydroalkoxycarbonylation of vinyl-estrone as a result of the vinyl-aromatic structural element. Similarly to terpenes, in the transformation of vinyl-estrone the determining factor is the internal asymmetric induction of the substrate, the chiral catalyst having a subordinate role.
4. Analytical method (MS, NMR techniques) has been elaborated for the rapid and exact determination of the deuterated species resulted in deuterioalkoxycarbonylation of styrene in presence of Pd(PPh3)2Cl2/SnCl2 catalyst precursors.
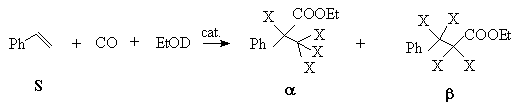
where X= H or D
5. On hand of the results obtained, the "hydride" mechanism has been supposed as being dominant under the reaction conditions applied.
(a) It has been shown, that isotope insertion is a result of reversible olefin insertions/ß-hydride eliminations prior to carbonylation.
(b) The deuterium content of unconverted substrate confirms the reversibility of the reaction paths and suggests the dissociation of the palladium s-complexes.
(c) The formation of all the deuterated species can be accomodated in terms of the reaction pathways supposed.
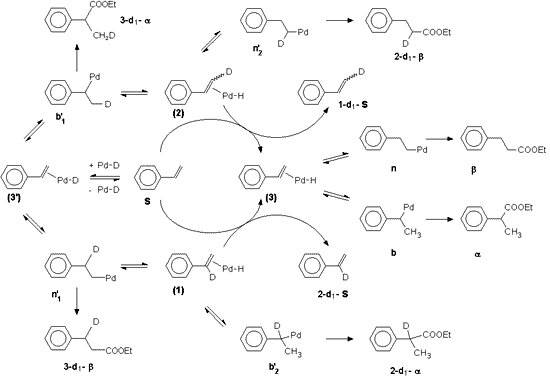
6. The marked difference in regioselectivity of hydroalkoxioxycarbonylation of styrene in the presence of the Pd(PPh3)2Cl2 and Pd(PPh3)2Cl2/SnCl2 catalyst systems is determined by the different behaviour of the metal-alkyl intermediates.
(a) In presence of Pd(PPh3)2Cl2 anti-Markownikoff addition of the palladium complex is inhibited by concurrent electronic factors (the polarization caused by the electron with-drawing effect of the phenyl ring, the p-benzylic interaction of the phenyl ring with the palladium), so the branched-chain product dominates. The reaction routes leading to the linear Pd-alkyl complex are irreversible.
(b) If SnCl2 is present, probably SnCl3- as ligand is formed, providing a sterically particularly hindered catalyst, which compensates the unfavourable electronic effects. In this system the transformations of the metal-alkyl intermediates are overall reversible.
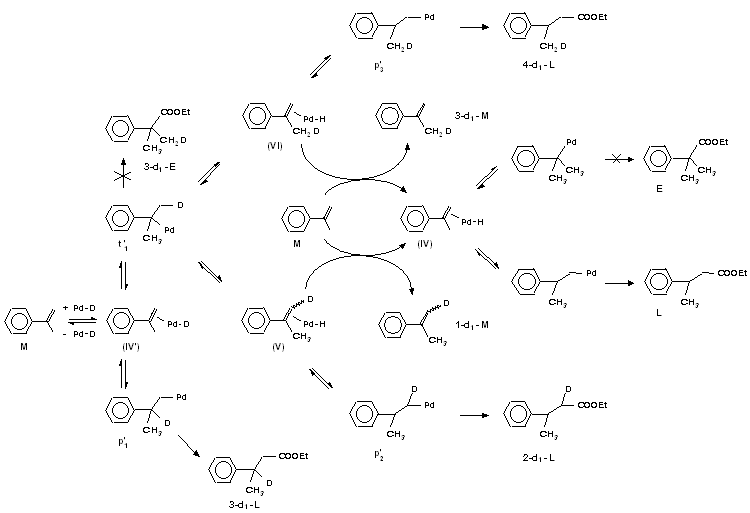
7. It has been established that a tertiary Pd-s-complex is formed during hydroalkoxycarbonylation of a -methyl-styrene, a species earlier not detected.
8. When this substrate is hydroalkoxycarbonylated, the significant difference in regioselectivity compared to styrene has been proved to be determined by the fast ß-elimination of the originally favoured tertiary Pd-alkyl. The isomerization process results in the dominant formation of the linear ester. 9. By means of a suitable method (chemical ionization) it has been possible to determine mass-spectrometrically the products formed in deuterioalkoxycarbonylation of 1-hexene. The application of MS and NMR techniques enabled the investigation of 1-hexene deuterioalkoxycarbonylation.
10. It has been proved that in the case of alkyl-olefins in presence
of Pd(PPh3)2Cl2 the terminal metal-alkyl
intermediate doesn't take part in the elimination processes, which explains
the observed decay in regioselectivity towards the branched product by
enhancing the reaction temperature. If SnCl2 is also present,
significant isomerization of the linear s-complex
takes place, resulting in a decrease of the amount of the linear ester
when the reaction conditions become harsher.
IV. Significance of the scientific results
Although the work belongs to basic research, the new products prepared
serve as possible building blocks for further functionalization or can
be regarded as potential intermediates in drugs syntheses. A better understanding
of the mechanism of the palladium catalyzed hidroalkoxycarbonylation gains
importance in the development of new, selective catalyst systems and the
designing of the reaction parameters. The intermediates derived from different
model compounds via deuterization and the reaction routes supposed proved
some new elements of the reaction mechanism, explaining thus the different
regioselectivities obtained in presence of the catalyst systems investigated.
Publications
1. Cs. Benedek, G. Szalontai, Á. Gömöry, S. Tõrös,
B. Heil: A Study on the Catalytic Pathways of the Hydroalkoxycarbonylation
Reaction of Styrene J. Organomet. Chem., 579 (1999) 145-155.
2. Cs. Benedek, S. Tõrös, B. Heil: Comparative Investigation
of Regioselectivity in Styrene and a -Methylstyrene Hydroalkoxycarbonylation
as a Function of Palladium Catalyst Structure, J. Organomet. Chem.,
586
(1999) 85-93.
3. Cs. Benedek, L. Prókai, S. Tõrös, B. Heil: Diastereoselective
Hydroalkoxycarbonylation of Terpenes and Vinyl-estrone, J. Mol. Catal.
A: Chem., 165 (2001) 15-21.
4. Cs. Benedek, Á. Gömöry, B. Heil, S. Tõrös:
Reversibility of Palladium-Alkyl Intermediate Formation in Deuterioalkoxycarbonylation
of 1-Hexene J. Organomet. Chem., 622 (2001) 112-120.
| Vissza a tartalomjegyzékhez
Back to Contents |
http://www.kfki.hu/chemonet/
http://www.chemonet.hu/ |