Vass Elemér
O–S(IV)–O és N–S(IV)–O axiális kötésrendszerû spiro-l4-szulfánok hidrolízisének mechanizmusa
Eötvös Loránd Tudományegyetem, Budapest
1997
A hipotetikus SH4 l4-szulfánoknak (régebben szulfuránoknak) nevezett tetraszubsztituált származékai a hipervalens kén(IV) vegyületek egy speciális családját képezik. E többé-kevésbé tökéletes trigonális bipiramis geometriájú vegyületekben az axiális kötések lényegesen gyengébbek az ekvatoriálisoknál, hipervalens axiális kötésrendszerük pedig igen könnyen polarizálható, aminek várhatóan hatása van a l4-szulfánok nukleofilekkel, pl. vízzel szembeni reaktivitására. Jóllehet az elmúlt évtizedek során számos aciklusos, mono- vagy spirociklusos l4-szulfánt állítottak elõ, a szakirodalomban alig található az ilyen típusú vegyületek nukleofil reakcióinak átfogó kinetikai vizsgálatával foglalkozó közlemény. Ezért kutatásaink a diaril(diaciloxi)- és diaril(acilamino)(aciloxi)spiro-l4-szulfánok szulfoxidokhoz vezetõ hidrolízis-mechanizmusának feltárására irányultak (ld. 1-2 irodalom). Az 1. sémában (sémákat ld. az angol nyelvû összefoglalóban) feltüntetett modellvegyületek e két osztálya az axiális ligandumok elektronegativitásában, s következésképpen az axiális kötésrendszer menti töltéseloszlásban különbözik. A reakciókat pszeudo-elsõrendû körülmények között, semleges és savas dioxán–víz elegyekben, valamint vizes puffer- vagy HClO4 oldatokban hajtottuk végre.
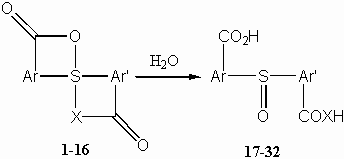
A kétegyenértékû S(IV)–O(acil) hipervalens axiális kötést tartalmazó diaril(diaciloxi)spiro-l4-szulfánok (1-8) bisz(karboxiaril)-szulfoxidokhoz(17-24) vezetõ hidrolízisérõl megállapítottuk, hogy a reakciót nagymértékben elõsegíti a nagy ionizációs erejû közeg, az aromás gyûrûn levõ elektronküldõ szubsztituensek növelik, az elektronvonzó szubsztituensek pedig csökkentik a reakciókészséget (a Hammett-egyenletbõl r = –0,52 reakciókonstans számítható). A reakció a szubsztituenstõl függõen többé-kevésbé erõsen savkatalizált (a katalitikus állandókra r kat = –1,55 érték adódik). Míg semleges közegben a reakció másodrendû deutérium oldószer-izotópeffektust mutat (kH2O/kD2O = 1,66), savas közegben erre rátevõdik a protonálódási elõegyensúly izotóphatása, így a savkatalizált reakcióknál inverz izotóphatást tapasztalunk (a katalitikus állandók aránya 0,56). A két öttagú spirogyûrût tartalmazó1-5 diaril(diaciloxi)spiro-l4-szulfánok hidrolízissel szembeni stabilitása lényegesen nagyobb (az ún. öttagú gyûrû-effektus tapasztalható). E kísérleti tények a 2. sémában bemutatott többlépéses reakciómechanizmust támasztják alá. Ennek értelmében a spiro-l4-szulfán elõbb egyensúlyi gyûrûfelnyílással egy monociklusos szulfónium-karboxilát típusú reaktív I ikerionná alakul (savas közegben az I’ karboxi-szubsztituált monociklusos szulfóniumkation keletkezik), melynek szulfóniumközpontját a következõ lépésben (I® II ill. I’® II’) vízmolekula támadja, majd gyors protonátmenetek révén képzõdik a szulfoxid végtermék. Mind a szubsztituenshatás, mind pedig a gyûrûtagszám hatása a spirociklusok stabibilitásának, azaz a reaktív szulfónium-karboxilát intermedier(I) képzõdési készségének döntõ jelentõségével magyarázható.
A diaril(acilamino)(aciloxi)spiro-l4-szulfánok (9-16) esetében az infravörös spektroszkópiai adatok (összhangban a korábbi röntgendiffrakciós vizsgálatokkal) a kiindulási vegyületek erõsen dipoláris szerkezetét igazolják. E molekulákban a központi kénatom jelentõs pozitív, az aciloxi-oxigén pedig jelentõs negatív töltéssel rendelkezik, és míg az S–N kötés majdnem teljes értékû kovalens kötésnek tekinthetõ, addig a szemközti S---O kötés a gyenge hipervalens kötések kategóriájába esik. Diaciloxi-analógjaikoz viszonyítva a diaril(acilamino)(aciloxi)spiro-l4-szulfánok vízzel szemben sokkal kevésbé reaktívaknak bizonyultak. Ebben az esetben az oldószer polaritásának és az ionerõsségnek nincs jelentõs hatása a hidrolízis sebességére. A reakciót az elektronvonzó szubsztituensek segítik elõ mind semleges (r = 1,43), mind pedig savas közegben (r cat = 0,90). Az aciloxi-részlet protonálása révén az erõs savak csak mérsékelten gyorsítják a reakciót. 50:50 térfogatarányú dioxán–H2O(D2O) elegyben kH2O/kD2O = 3,68 elsõrendû deutérium oldószer-izotóphatás mérhetõ. A nitrogént öttagú gyûrûben tartalmazó 9-14 spiro-l4-szulfánok sokkal reaktívabbak hattagú gyûrûs analógjaiknál (15-16). A diaril(acilamino)(aciloxi)spiro-l4-szulfánok esetében a 3. sémában feltüntetett mechanizmus javasolható, mely feltételezi, hogy a sebességmeghatározó lépésben a víz nukleofil támadásával egyidejûleg az O–H kötés is felhasad, ugyanakkor az új S–O kötés kialakulása a III (savas közegben pedig a III’) átmeneti állapoton keresztül az S–N kötés jelentõs mértékû lazításával jár. A vízzel szemben egyébként kis reakciókészséget mutató, a nitrogént hattagú gyûrûben viselõ 15-16 spiro-l4-szulfánok hidrolízisekor az oldatban jelenlevõ OH–-ionokkal végbemegy egy gyors párhuzamos reakció, mely még semleges közegben sem hanyagolható el. Az (acilamino)(aciloxi)spiro-l4-szulfánok és diaciloxi-analógjaik közti reaktivitásbeli különbségek egyrészt az erõsen polarizált N–S(IV)–O axiális kötésrendszer disszimmetriájából, másrészt pedig az acilamino-részletnek az aciloxihoz viszonyított rosszabb távozócsoport-jellegébõl adódik.
A dolgozatban tárgyalt eredmények hozzájárulnak a hipervalens S(IV)-vegyületek kénközponton lejátszódó nukleofil szubsztitúciós reakcióinak jobb megértéséhez.
Mechanism of hydrolysis of spiro-l4-sulfanes with O–S(IV)–O and N–S(IV)–O axial bond system
Ph.D. thesis, Eötvös Loránd University,Budapest
1997
The tetrasubstituted derivatives of the hypothetical SH4 molecule known as l4-sulfanes (or sulfuranes) constitute a special class of hypervalent S(IV) compounds. In these compounds with more or less perfect trigonal bipyramidal geometry the axial bonds are significantly weaker than the equatorial ones and their hypervalent axial bond system is highly polarizable, which should be reflected in the reactivity of these compounds toward nucleophiles, e.g. water. Although a great number of acyclic, monocyclic and spirocyclic l4-sulfanes have been synthesised during the past decades, with a few exceptions no detailed kinetic studies on nucleophilic reactions of such compounds have been published. Therefore we have investigated the mechanism of hydrolysis of diaryl(diacyloxy)- and diaryl(acylamino)(acyloxy)spiro-l4-sulfanes leading to sulfoxides (see Refs. 1-2 and Scheme 1). These two classes of model compounds differ in the electronegativity of the axial ligands, and consequently in the charge distribution along the axial bond system. Kinetic measurements were performed under pseudo-first-order conditions in neutral and acidic dioxane-water mixtures, in aqueous buffer or HClO4 solutions.
| X=O | Ar | Ar’ | X=NMe | Ar | Ar’ |
| 1, 17 | 1,2-phenylene | 1,2-phenylene | 9, 25 | 1,2-phenylene | 1,2-phenylene |
| 2, 18 | 5-NO2-1,2-phenylene | “ | 10,26 | 5-NO2-1,2-phenylene | “ |
| 3, 19 | 5-Cl-1,2-phenylene | “ | 11, 27 | 5-Cl-1,2-phenylene | “ |
| 4, 20 | 5-MeO-1,2-phenylene | “ | 12, 28 | 5-Me-1,2-phenylene | “ |
| 5, 21 | 5-NMe2-1,2-phenylene | “ | 13, 29 | 5-MeO-1,2-phenylene | “ |
| 6, 22 | 1,8-naphtylene | “ | 14, 30 | 1,8-naphtylene | “ |
| 7, 23 | “ | 1,8-naphtylene | 15, 31 | 1,2-phenylene | 1,8-naphtylene |
| 8, 24 | 2,2’-biphenylilene | 1,2-phenylene | 16,32 | 1,8-naphtylene | “ |
Scheme 1
For the hydrolysis of diaryl(diacyloxy)spiro-l4-sulfanes
1-8 with two equivalent S(IV)–O(acyl) hypervalent axial bonds,
leading to bis(carboxyaryl)-sulfoxides 17-24 it has
been found that the hydrolysis is considerably enhanced by reaction media
with high ionising power, electron-releasing substituents on the aromatic
ring increase, whereas electron-withdrawing ones decrease the reactivity
(from the Hammett equation acatalytic constant r=–0.52
was obtained). The reaction is more or less strongly catalysed by acids,
depending on the substituent (for the catalytic constants rcat=–1.55).
In neutral media the reaction shows a secondary deuterium solvent isotope
effect (kH2O/kD2O = 1.66). In acidic
media this is superimposed on the isotope effect of the protonation pre-equilibrium,
and therefore an inverse global isotope effect is observed (the ratio of
catalytic constants was 0.56). Diaryl(diacyloxy)spiro-l4-sulfanes1-5
containing two five-membered spirorings are significantly more stable towards
hydrolysis (the so-called five-membered ring effect has been observed).
On the basis of these findings a multi-step mechanism has been proposed
(Scheme 2), according to which the spiro-l4-sulfane
in solution undergoes first an equilibrium process like valence isomerization
to give a reactive monocyclic sulfonium-carboxylate zwitterion I (in
acidic media protonation leeds to a carboxy-substituted monocyclic sulfonium
cation I’); the sulfonium species, attacked in the following step
(I® II or I’®
II’) by a water molecule, is converted finally into sulfoxide by fast
proton-transfer steps. Both the substituent effect and the ring-size effect
observed have been correlated with the stability of the spirorings, i.e.
with the readiness of the formation of a reactive sulfonium-carboxylate
intermediate (I).
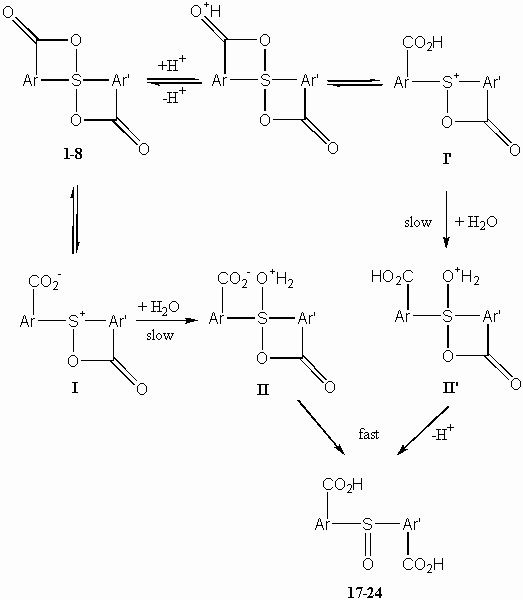
Scheme 2
For diaryl(acylamino)(acyloxy)spiro-l4-sulfanes (9-16) IR spectroscopic data (in agreement with previous X-ray studies) support the dipolar structure of the starting compounds in which the central sulfur and the acyloxy-oxygen atoms bear considerable positive and negative charge, respectively. In these compounds the S–N bonds can be regarded as almost full-valence covalent bonds, while the opposite S---O bonds fall inthe category of weak hypervalent bonds. Diaryl(acylamino)(acyloxy)spiro-l4-sulfanes proved to be much less reactive towards water than their diacyloxy analogues. In this case solvent polarity and ionic strength had no significant influence on the rate of hydrolysis. The reaction is promoted by electron-withdrawing substituents in both neutral (r = 1.43) and acidic media (r cat = 0.90). Strong acids accelerate the reaction moderately by protonating the acyloxy group of the substrate. In 50:50 (v/v) dioxane–H2O(D2O) the primary deuterium isotope effect is kH2O/kD2O = 3.68. Spiro-l4-sulfanes 9-14 with five-membered N-containing ring are much more reactive than their six-membered analogues 15-16. For diaryl(acylamino)(acyloxy)spiro-l4-sulfanes a mechanism involving the nucleophilic attack of water on the sulfonium centre with simultaneous O–H bond cleavage in the rate-determining step is proposed (Scheme 3). The formation of the new S–O bond in the rate determining step (through transition states III in neutral and III’ in acidic media, respectively) occurs with a considerable loosening of the S–N bond. Spiro-l4-sulfanes with six-membered N-containing spirorings, which are slightly reactive towards water, undergo a fast parallel reaction with OH– ions even in neutral solutions. The differences in the reactivity of (acylamino)(acyloxy)spiro-l4-sulfanes relative to their diacyloxy analogues can be attributed to the disproportion of bond strengths in the highly polarized axial N–S(IV)–O array, as well as to the worst leaving group character of the acylamino moiety.
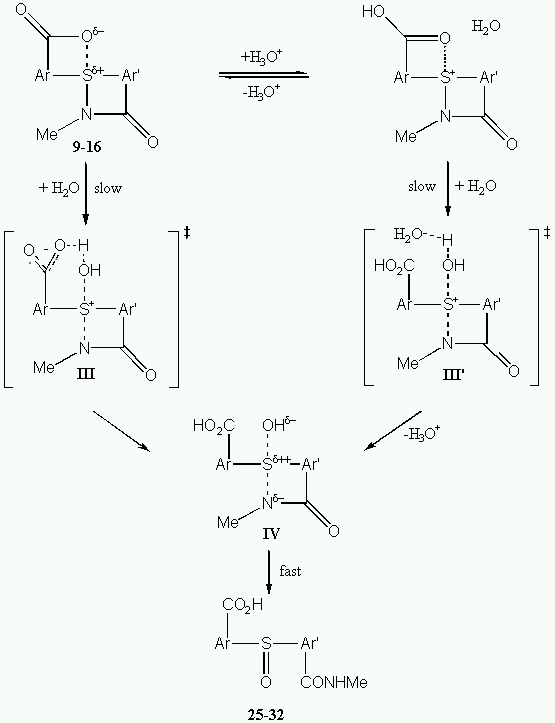
Scheme3
The results presented in this work contribute to the better understanding of the nucleophilic substitution reactions occuring at sulfur atom in hypervalent S(IV) compounds.
Publications
E.Vass, F. Ruff, I. Kapovits, J. Rábai and D. Szabó: "Diaryldiacyloxyspirosulfuranes. Part 4. A kinetic study on the mechanismof hydrolysis", J. Chem. Soc. Perkin Trans.2, (1993) 855-859.
E.Vass, F. Ruff, I. Kapovits, D. Szabó and Á. Kucsman: "Spiro-l 4-sulfanes withN-SIV-O axial bond system.
A kinetic study on the mechanism of hydrolysis", J. Chem. Soc. Perkin Trans. 2, (1997) 2061-2068.
| Vissza a tartalomjegyzékhez Back to Contents |
Vissza a PhD-tézisek tartalomjegyzékhez Back to PhD theses list |
http://www.kfki.hu/chemonet/ http://www.ch.bme.hu/chemonet/ |