Metastabil ionok tanulmányozása tömegspektrometriai módszerekkel
Eötvös Loránd Tudományegyetem, Budapest
1998
A metastabil ionokkal kapcsolatos elsõ megfigyeléseket több mint ötven évvel ezelõtt publikálták és kezdetben a metastabil ionok vizsgálata a tömegspektrometria különleges területének számított, azonban napjainkban világszerte számos tömegspektromteriai laboratórium fõ kutatási profilja a gázfázisban lejátszódó unimolekuláris bomlások vizsgálata. A metastabil ionok vizsgálatával olyan információk nyerhetõk az adott bomlásfolyamatról, amelyek más kísérleti módszerrel nem határozhatók meg.
A doktori disszertációmban mely az MTA Kémiai
Kutatóközpont Kémiai Intézet Tömegspektrometriai
Csoportjában készült többféle módszerrel
vizsgáltam a metastabil bomlásfolyamatokat, így az
három fejezetre osztható. Az elsõ fejezet egy módszer
kifejlesztését ismerteti, a második a módszer
alkalmazására mutat be egy példát, a harmadik
fejezet témája pedig a metastabil bomlásfolyamatok
energiafüggésének vizsgálata MALDI ionizáció
esetében.
1. A kinetikus energiafelszabadulás eloszlásfüggvényének meghatározása
Metastabil módon bomlanak a tömegspektrométerben azok az ionok, amelyek a gyorsítás után de még a detektálás elõtt fragmentálódnak. Tömegközépponti koordinátarendszerben szemlélve egy elemi bomlásfolyamatot az egy irányultsággal rendelkezik. Attól függõen, hogy a bomlás irányultsága milyen, a fragmensek sebessége csökken vagy növekszik a prekurzor ion sebességéhez viszonyítva. Ez az effektus a tömegspektrumban megjelenõ csúcsok kiszélesedését eredményezi. A kiszélesedés mértéke attól függ, hogy
a bomlás során keletkezett fragmensek milyen sebességgel távolodnak el egymástól, vagyis attól, hogy a folyamat során kinetikus energia formájában milyen nagyságú energia szabadul fel. A csúcsalak analízisével információ nyerhetõ a kinetikus energiafelszabadulásról (KER kinetic energy release), amely a bomlásfolyamatot jellemzõ potenciális energiaprofillal van kapcsolatban.
A kinetikus energiafelszabadulást legtöbbször egy diszkrét értékkel adják meg annak ellenére, hogy egy eloszlásfüggvény (KERD kinetic energy release distribution) nyilvánvalóan sokkal pontosabban jellemezheti az adott folyamatot. Ennek a legfõbb oka az, hogy a KERD függvény meghatározása a kísérleti csúcsprofilból csak bonyolult számításokkal lehetséges. Erre a célra már számos módszert kidolgoztak, azonban egyik sem áll rendelkezésre olyan formában, amely egyszerûen alkalmazható. Célom ezért az volt, hogy egy olyan könnyen használható számítógépes programot dolgozzak ki, amely egy metastabil csúcsprofilból a folyamatot jellemzõ KERD függvényt határozza meg
A kifejlesztett algoritmus részletes leírását az értekezésen kívül egy referált folyóiratban megjelent publikáció is tartalmazza, így itt csak a program fõbb jellemzõit foglalom össze:
2. Többszörösen protonált peptidek gázfázisú konformációjának vizsgálataA program alkalmazásához csak a tömegspektrométer könnyen hozzáférhetõ jellemzõi szükségesek.Különbözõ csúcstípusok esetén is használható (Gauss-típusú vagy ahhoz hasonló csúcsprofilok, nagy kinetikus energiafelszabadulás esetén megjelenõ dish-topped csúcsok, stb.).
A program a tömegspektrométer diszkriminációs effektusát (nagy kinetikus energiafelszabadulás esetén a fragmensek egy része nem detektálható) a kollektor elõtti szûkítõ rés méretével ill. az elektrosztatikus analizátor ún. határszöge alapján számítja.
Figyelembe veszi a tömegspektrométer csúcstorzító hatását, ami különösen fontos igen kis kinetikus energiafelszabadulások esetén.
A kinetikus energia felszabadulást közvetlenül a csúcsprofilból számítja, így valamilyen simító módszert legtöbbször alkalmazni kell.
Régóta ismert, hogy az ún. töltésszeparációs metastabil folyamatok esetében amelyekben többszörös a töltésû ionból két töltéssel rendelkezõ termék keletkezik a kinetikus energiafelszabadulás szinte kizárólag a termékionok közötti elektrosztatikus taszításból származik. Ez azt jelenti, hogy ha meg tudjuk határozni a kinetikus energiafelszabadulást, akkor információt nyerhetünk a töltéscentrumok távolságáról is.
Néhány referenciavegyület esetében a metastabil
csúcsprofilból számított KERD függvény
átlagértékének (T) segítségével
határoztam meg a töltések távolságát
(r), a következõ összefüggés felhasználásával:
r
= c / T (ahol c konstans). A referenciavegyületek
vizsgálatának célja egyrészt annak alátámasztása,
hogy az elõbbi összefüggéssel számított
adatok összhangban vannak a más módszerekkel nyert töltéstávolságokkal,
másrészt annak demonstrálása, hogy a közelítés
alkalmazható 1 nm-nél nagyobb töltéstávolságok
esetén is. Az 1. táblázat adatai mindkét feltevés
érvényességét igazolják.
1. táblázat
A KERD függvények alapján számított töltéstávolságok összehasonlítása más módszerekkel meghatározott adatokkal a referencia vegyületek töltésszeparációs folyamatainak esetében. A * az irodalomból vett adatokat jelöli.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A töltéstávolságok meghatározásán alapuló módszert peptidek gázfázisban kialakult konformációjának vizsgálatára alkalmaztam. A többszörösen protonált peptidek gázfázisú konformációja régóta vitatott kérdés, ugyanis kezdetben kézenfekvõnek tûnt az az elképzelés, amely szerint a töltések közötti nagymértékû elektrosztatikus taszítás eredményeképpen a kinyújtott konformáció a preferált. Az utóbbi években publikált kísérleti tapasztalatok azonban bizonyos esetekben éppen ennek az ellenkezõjére mutatnak rá.
Az elõbbiekben bemutatott módszer akkor lehet alkalmas egy peptid konformációjának vizsgálatára, ha hasonlóan a referenciavegyületekhez a töltéscentrumok helye jól meghatározható. A bradykinin (RPPGFSPFR) esetében ez egyértelmû, hiszen mindkét terminális a legnagyobb gázfázisú bázicitással rendelkezõ arginin. A melittin (GIGAVLKVLTTGLPALISWIKRKRQQ-NH2) esetében hasonlóan az aminosavak relatív bázicitása határozza meg a protonálódást, így a háromszorosan protonált molekula nagy valószínûséggel a 7K, 22R és 24R aminosavakon protonálódik. A kétszeresen protonált trilizin ilyen szempontból már sokkal bonyolultabb rendszer, mert a három közel azonos bázicitású lizin oldalláncon a két proton különbözõ kombinációkban helyezkedhet el, továbbá az oldalláncok flexibilitása is befolyásolhatja a töltések távolságát.
A bradykinin és a melittin esetében a vizsgált
töltésszeparációs folyamatoknak megfelelõ
csúcsprofilból meghatározott KERD függvény
átlagértékei alapján számított
töltéstávolságok sokkal kisebbeknek adódtak,
mint a teljesen kinyújtott konformációnak megfelelõ
töltéstávolság. A KERD föggvények
további jellemzõje, hogy hasonló szélességûek,
mint a referenciavegyületek esetében kapott eloszlásfüggvények,
amely arra utal, hogy az említett két (többszörösen
protonált) peptid a gázfázisban a nagy elektrosztatikus
taszítás ellenére sem egy kinyújtott, hanem
egy globuláris jellegû konformációval rendelkezik.
Ez összhangban van a melittin szilárd fázisú
a
-helikális szerkezetével is. A trilizin esetében kapott
KERD függvény az elõzõeknél szélesebb,
amely magyarázható a protonok elhelyezkedésének
különbözõ kombinációival, de az oldalláncok
flexibilitásával is. Az eredmények alapján
nyilvánvaló, hogy a kinyújtott konformáció
még egy ilyen flexibilis rendszernél is csak igen csekély
valószínûséggel alakul ki. Az említett
peptidek vizsgálatával kapcsolatos eredményeket a
2. táblázat foglalja össze.
2. táblázat
A töltésszeparációs. A KERD függvények alapján számított töltéstávolságok összehasonlítása a teljesen kinyújtott konformációnak megfelelõ értékekkel a többszörösen protonált peptidek töltésszeparációs folyamatainak esetében. (A melittin esetében az 22R és 24R oldalláncokon lévõ protonoknak megfelelõ töltéscentrumot vettem figyelembe.)
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
||
|
|
||
|
|
|
|
3. MALDI-PSD spektrumok lézerenergia-függésének vizsgálata
A fragmensek képzõdését számos kísérleti paraméter befolyásolja és azok közül a legtöbbnek a hatását részletesen vizsgálták eddig. A MALDI-PSD spektrumok függése a lézer energiájától azonban nem volt megfelelõen dokumentálva az irodalomban, ezért célom annak vizsgálata volt.
A modellvegyületek egy 22 aminosavból álló peptid ill. egy triszacharid voltak. Mindkét vegyület esetében három különbözõ, konstans lézerenergián regisztrált spektrumot hasonlítottam össze. Alacsony lézerenergián csak igen szelektív, fõként a fragmensek felsõ tömegtartományára korlátozódó fragmentáció volt megfigyelhetõ. Ez a vizsgált peptid esetében az intramolekuláris kölcsönhatásokkal (sóhidak képzõdése) magyarázható. A peptid különbözõ lézerenergián regisztrált PSD spektrumaiban néhány prekurzor-fragmens ionpár esetében a intenzitásarányok monoton változását mutattam ki, amely azt támasztja alá, hogy a különbözõ fragmentációs utak eltérõ lézerenergiát igényelnek.
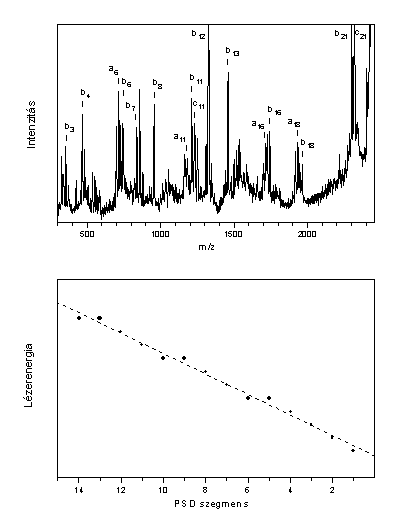
Kísérleti adataim alapján megállapítottam, hogy peptidek esetében az alacsony, konstans lézerenergia alkalmazása a sóhidak képzõdésével végbemenõ szelektív fragmentáció révén a savas aminosav egységek lokalizálásában lehet hasznos. Rámutattam továbbá a lézerenergia kontrollált módon történõ változtatásának fontosságára is. A vizsgált peptid esetében a lézerenergiának (az egyes spektrumszegmensek számának függvényében történõ) lineáris változtatásával regisztrált PSD spektrum tartalmazza a legtöbb karakterisztikus fragmenst, ugyanakkor azok relatív intenzitása is összehasonlítható. A peptid így regisztrált MALDI-PSD spektruma és a lézerenergia változása az 1. ábrán látható.
1. ábra
Investigation of metastable ions by mass spectrometry
Ph.D. thesis, Eötvös Loránd University,
Budapest
1998
The first observations in the field of mass spectrometry were published more than fifty years ago. Initially the investigation of metastable ions was considered as a exotic area of mass spectrometry. Today, however, there are numerous mass spectrometry research groups which are mainly interested in unimolecular gas phase decompositions. This is not surprising, because the study of metastable decompositions provides data about a given system which are not, or not easily available by other experimental techniques.
In the Mass Spectrometry Group, Institute of Chemistry, Chemical Research
Center, Hungarian Academy of Sciences
I have studied the metastable decompositions by various methods. The
results are summarised in three chapters in this thesis. The development
of a method can be used for evaluation of experimental data is described
in the first chapter; the second one presents an example for the application
of the developed method; and the laser power dependence of MALDI-TOF PSD
(matrix-assisted laser desorption/ionization time-of-flight post-source
decay) spectra is discussed in chapter three.
1. Determination of kinetic energy release distributions
Metastable ions "survive" the acceleration but dissociate spontaneously before they reach the ion detector. In center-of-mass co-ordinate system the metastable decomposition has an orientation. Depending on their orientation the velocity of the product ions become larger or smaller than that of the precursor ion. This effect can be observed as peak broadening in the recorded mass spectra. The degree of the peak broadening depends on the difference of the velocity of the product particles, in other words on the kinetic energy released in the decomposition. Therefore, the analysis of the metastable peak profile can provide information about the kinetic energy release and the potential energy profile of the decomposition as well.
Although the kinetic energy release can be described much more accurately by a distribution function it is often characterised by a discrete (average) value. The main reason for this is that the kinetic energy release distribution (KERD) can only be calculated by use of complicated algorithms. Various methods have been developed and described in the literature, however those are rather complicated and are not easy to use. The primary aim of my work has been the development of a computer program that can easily be applied for calculation of KERD from experimental metastable peak profile.
A detailed description of the developed algorithm can be found in my thesis and it has been published [ref. 1], here only its basic features are summarised:
2. Investigation of gas phase conformation of multiply protonated peptidesa) The program requires only the parameters of the mass spectrometer that are readily available;b) It can be used for analysis of various metastable peak profiles (Gaussian-type, "dish-topped", etc.);
c) The discrimination effect of the mass spectrometer (in case of large kinetic energy release a given fraction of the fragments can not be detected due to their large angular divergence) is taken into account by the width of the collector slit and the acceptance angle of the electrostatic analyser;
d) The algorithm can take into account the distortion effects of the mass spectrometer, that is particularly important in case of small kinetic energy releases;
e) It calculates the KERD directly from the experimental peak profile, so it is often necessary to use a smoothing step prior to the calculation of KERD.
In case of a charge separation decomposition when two charged particle form from a multiply charged ion the kinetic energy release is mainly determined by the electrostatic repulsion between the two product ions. Therefore, if the kinetic energy release can be experimentally determined then the distance between the charges can also be characterised (in the transitional state).
In case of some reference systems the average value (T) of the
KERD determined from experimental metastable peak shape was used for calculation
of intercharge distance (r), using the following simple equation:
r
= c / T (where c is a constant). The aim of these
calculations was to check that if the data calculated by this approach
are consistent with those determined by other methods, on the other hand
to demonstrate the applicability of our approach in the case of intercharge
distances larger than 1 nm. The data shown in Table 1 support the validity
of both assumptions.
Table 1
Comparison of intercharge distances calculated from KERD and using other methods, in case of the investigated reference systems. (* shows literature data.)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The developed approach was applied for investigation of conformation of multiply protonated peptides in the gas phase. This has been a controversial issue for long time. Initially, it assumed that owing to the large electrostatic repulsion between the charges the multiply protonated peptides have an extended conformation. However, the experimental data obtained in the last few years suggest the contrary.
The method based on the determination of intercharge distances is suitable
for investigation of the conformation
of peptides if the charges (protonation sites) can be localised.
In the case of doubly protonated bradykinin (RPPGFSPFR)
the protonation sites are unambiguous, because both termini
are arginin residues with the largest gas phase basicity. Similarly
in the case of triply protonated melittin (GIGAVLKVLTTGLPALISWIKRKRQQ-NH2)
the protonation sites were determined by the relative gas phase basicity,
thus the most probable protonated residues are: 7K, 22R
and 24R. From the same point of view the doubly protonated trilysin
is a more complicated system, because the three side chains have very similar
gas phase basicity. So the two protons may be localised in various combinations,
moreover the flexibility of the side chains can also affect the distance
of the protons.
The intercharge distances calculated from the average values of KERD
were much smaller than those corresponding to the extended conformation,
for all three peptides. The width of the KERDs in case of bradykinin and
melittin were very similar to those obtained for the reference systems,
which indicates that the two peptides have a "rigid" conformation. This
is in a good agreement with that the melittin has a-helical
conformation in the solid phase. The KERD obtained for trilysin is wider
than the others, which can be explained by the flexibility of the molecule
and the three alternative protonation sites. However, in spite of its small
size and flexibility the extended conformation of the double protonated
molecule is very unlikely. The data obtained on the conformation of multiply
protonated peptides are summarised in Table 2.
Table 2
Comparison of intercharge distances calculated from KERDs and those
corresponding to extended conformations, in case of multiply protonated
peptides. (For melittin the charge center of 22R and 24R
side chains has been taken into account.)
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
||
|
|
||
|
|
|
|
3. Investigation of laser power dependence of MALDI-PSD spectra
The fragmentation of molecular ions depends on numerous experimental parameters (pressure of residual gas, etc.) in case of MALDI-PSD spectra, and most of them have been studied in detail. The effect of laser power, however, has not been thoroughly studied.
In this work a peptide (consisting of 22 amino acid residues) and a trisaccharide was used as model compounds. In both cases three PSD spectra were recorded at different, constant laser powers, and those were compared to each other. At low laser power a selective fragmentation was only observed, and the fragments appeared mainly in the high mass region. In case of the peptide this can be explained by intramolecular interactions (formation of salt bridges). In the PSD spectra of the peptide recorded at different laser powers it was found that the intensity ratio of some selected ion-pairs increases with the
laser power. This phenomenon can be explained by that different fragmentation pathways require different activation energy.
On the basis of the experimental results it was concluded that low laser power in MALDI may be useful in localisation of acidic side chain residues. However, if the purpose of the PSD analysis is identification of as many fragments as possible (e.g. sequencing), the laser power should be increased as a linear function of the number of PSD segments. (The applied MALDI-TOF instrument has a two-stage reflectron, which focuses the fragments only in a small mass range, thus the whole PSD spectra were obtained from several spectrum segments.) A PSD spectrum recorded in this way is shown in Figure 1.
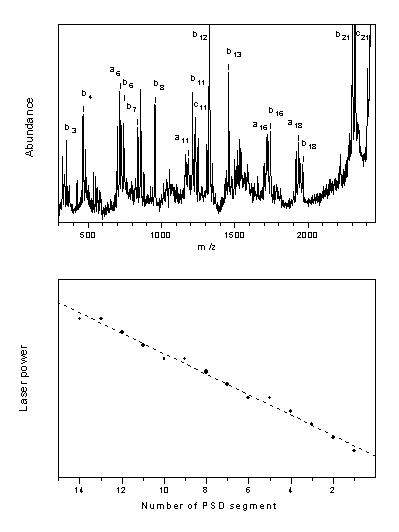
Figure 1
MALDI-PSD spectrum of ACTH(18-39) peptide recorded at "optimised" laser
power
Publications
1. Zoltán Szilágyi, Károly Vékey: A simple algorithm for the calculation of the kinetic energy release distribution. Eur. Mass Spectrom. 1 (1995) 507-5182. Zoltán Szilágyi, László Drahos, Károly Vékey: Conformation of doubly protonated peptides studied by charge-separation reactions in mass spectrometry. J. Mass Spectrom. 32 (1997) 689-696
3. Károly Vékey, Zoltán Szilágyi: Kinetic energy release and intercharge distance of the protonitronium dication (HONO2+). Int. J. Mass Spectrom. Ion Proc. 165/166 (1997) 1-4
4. Zoltán Szilágyi, Julie E. Varney, Peter J. Derrick, Károly Vékey: Dependence of matrix-assisted laser desorption/ionization post-source decay spectra on laser power. Rapid Commun. Mass Spectrom. 12 (1998) 489-492
| Vissza a tartalomjegyzékhez
Back to Contents |
Vissza a PhD-tézisek tartalomjegyzékhez
Back to PhD theses list |
http://www.kfki.hu/chemonet/
http://www.ch.bme.hu/chemonet/ |