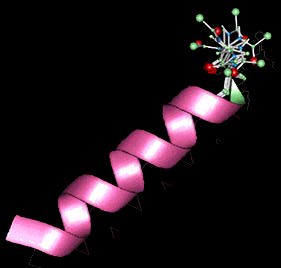
Ez az ábra, amely Brooks és Young egy hetero-CHARMM számításának az eredménye, egy alfa hélixet mutat (rózsaszínû szalag) a láncot lezáró aminosav hat különféle helyzetével. Az aminosavat egy golyókból és rudacskákból álló alakzat reprezentálja, ahol az oxigén piros, a szén zöld, a nitrogén atomok pedig a kék golyók.
A számítások rámutattak arra, hogy a hélix-képzôdésben "együttmûködés" tapasztalható, azaz a molekula egyik részének a megváltozása más részekre is hatással van. A hat egymásra vetített struktúra az "esernyô-mintavételezés" nevû számítási technika eredménye: a számításokat egy olyan konfiguráción végzik, amely a vizsgált alakzat lehetséges szabadenergia-terében van kifeszítve. Az aminosav legkülsô szénatomjának hat különbözô helyzete 360 fokos ívet alkot, amint hidrogénkötést létesítve visszafordul a hélix felé.
Charles Brooks és a Pittsburgh Supercomputing Center kutatója, Bill Young együttmûködtek abban, hogy fehérje-szerkezeti kutatásaikban minél jobban kiaknázhassák a T3D párhuzamos munkavégzô képességét. Az általuk kifejlesztett módszer a CHARMM (Chemistry at HARvard Macromolecular Mechanics) egy speciális változata. A CHARMM – amelyet 15 évvel ezelôtt Martin Karplus, Brooks és mások dolgoztak ki – keletkezése óta az egyik leginkább használt eszközzé vált a fehérjekutatásban és a gyógyszer-fejlesztésben.
Néhány évvel ezelôtt Brooks és Young kidolgoztak egy módszert, amelynek segítségével képesek megosztani az MD-számításokat párhuzamos rendszerek – például a Connection Machine, CM-2 vagy CM-5 – és hagyományos vektor-rendszerû szuperszámítógépek között, mint amilyen a CRAY Y-MP vagy C90. A CHARMM-nak ez az általuk kidolgozott heterogén (azaz két különbözô rendszeren futó) változata képes megfelelni azoknak az elvárásoknak, amelyek elé például a vizes közegben oldott fehérjék szimulációja állítja. Az ô megközelítésük azon alapul, hogy a számítások nagy része a vízmolekulák közötti kölcsönhatás leírásához szükséges, és ezek a kölcsönhatások eredendôen párhuzamosak -- azaz az egyes molekulákra vonatkozó számítások függetlenek a többiektôl. A munka ezen része nagyon hatékonyan elvégezhetô párhuzamos rendszereken, míg ezzel egy idôben más effektusok – azaz a fehérje önmagára gyakorolt hatása, ill. a víz-fehérje kölcsönhatás – a vektor-rendszerû gépen számíthatók.
A CHARMM-nak ez a megosztott változata igen sikeres, elsôdlegesen a CRAY T3D képességeinek köszönhetôen, ámbár a különbözô számítógépek közti kommunikáció és más technikai problémák ronthatják a teljesítményt. A T3D-vel és a C90-nel Brooks és Young sok technikai problémát megoldott. Egy tesztben, amelyben különféle szerkezeteket vizsgáltak, a hetero-CHARMM a 32 T3D processzort használó T3D/C90 pároson két-háromszor olyan gyorsan mûködött, mint a C90-en egymagában.
"A molekuladinamika ideális terület a párhuzamos számítógépek számára, a T3D jóval könnyebben alkalmazható, a processzorok pedig gyorsabbak, mint más, általunk eddig használt rendszerben" – állítja Young. "Különösen akkor növekszik látványosan a teljesítmény, ha a processzorok számát megnöveljük. Az általunk készített algoritmus egyenlôen tudja megosztani a munkát a processzorok között, igy sokkal jobban ki tudjuk használni a párhuzamos számítógépet és jóval gyorsabbak lehetünk."
Young az alfa-hélix (a fehérje-struktúra egy alapmintája) kutatásainak felgyorsítására kezdte alkalmazni a CHARMM-ot a T3D/C90 pároson. "Elégedett vagyok" – mondta Young – "hogy végre túl vagyunk a technikai problémákon és most már élesben futtathatjuk a programokat, azaz ténylegesen a tudománnyal foglalkozunk." Az ô számításai azt a termodinamikai változást vizsgálják, amely akkor következik be, amikor a hélix elkezd egy újabb csavarodást.