Antitumor hatású molekulák
Jeltovábbítás-gátlás és programozott sejthalál-indukálás
A tumoros transzformáció és a sejtek közötti kommunikáció
Az elmúlt évek során alapvetô áttörés következett be a tumorképzôdés és -kifejlôdés molekuláris biológiai értelmezésében. Mai ismereteink szerint a rosszindulatú daganatsejtek kialakulása többlépcsôs folyamat következménye, mely egyrészt genomiális változások sorozatára (többek közt bizonyos onkogének aktivációjára, illetve szupresszor gének inaktivációjára), másrészt az intercelluláris kommunikációs rendszer rendellenes mûködésére vezethetô vissza (1. ábra). A rosszindulatú daganatsejt, azaz a tumoros transzformáció létrejöttének elsô lépései olyan genomiális változásokból állnak, melyek folyamatos sejtosztódáshoz vezetnek. Különbözô rákkeltô kémiai anyagok, radioaktív és ultraibolya sugárzás és egyéb környezeti ártalmak génkárosító, azaz mutagenikus hatásuk révén nagymértékben fokozzák annak valószínûségét, hogy elõnyös sejtosztódási tulajdonságokkal rendelkezõ transzformált sejtek jöjjenek létre, amit a DNS repair rendszer sérülései, illetve a tumor szupresszor gének mutációi tovább erôsítenek. Az onkogének és tumor szupresszor gének nagyszámú, különbözô funkciójú proteint kódolnak. Az onkogének különbözô hatásokra bekövetkezô aktivációja és a tumor szupresszor gének inaktivációja folyamatos proliferációs jelet generálnak és kiiktatják, illetve gátolják a sejtciklust kontrolláló fehérjék mûkôdését.
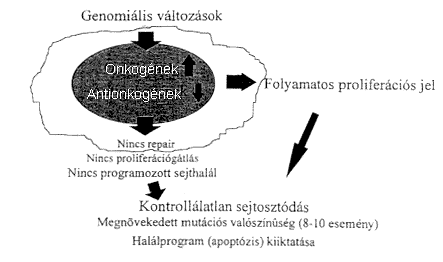
1. ábra. A tumorsejt kialakulása
A normál szervezetben a sejtosztódás és -differenciálódás folyamatai, illetve a specializálódott sejtek mûködése igen nagyfokú szabályozottságot mutatnak, és több károsító tényezõ együttes fönnállása szükséges ennek a szabályozási folyamatnak a kikapcsolásához. Ez a szabályozottság nemcsak a sejt belsô genetikai programjától, hanem a sejtek közötti kommunikációtól is függ. Az emberi szervezetben a sejtek igen magas szinten szervezett és kommunikáló sejttársadalomban élnek, ahol a társadalom egészének, illetve egyes részeinek zavarai tükrözôdnek az egyes sejtek életében és "sorsában", és az egyéni sejt mûködésének totális zavara végzetes lehet az egész szervezetre nézve. Az egy mindenkiért, mindenki egyért elve a sejttársadalomban sokkal jobban érvényesül, mint az emberi társadalomban, ami többek közt az egyéni sejt önfeláldozó és a sejttársadalom védelmében elkövetett öngyilkossági képességében (az apoptózisban) is megnyilvánul. A cseppben a tenger és a tengerben a csepp filozofikus gondolatát, illetve a holografikusan fölépülô sejttársadalom elvét a legújabb kutatások a központi idegrendszer, illetve a memória mûködésében is egyértelmûen kimutatták. A holografikus képet (illetve pl. a memóriát) az jellemzi, hogy a rendszer egészére vonatkozó kép a rendszer egyes elemeiben is megtalálható, de a kép, illetve a jel lokalizációja a kép élességének rovására megy. A tumorok többnyire monoklonális eredetûek, azaz egyetlen transzformálódott sejtbôl alakulnak ki, de ebben a szervezetre nézve negatív evolúciós folyamatban a rendszer egészének hibái, illetve fatális kommunikációs zavarai tükrözôdnek.
A tumor kialakulása során a szaporodást és differenciálódást szabályozó folyamatok amelyek normális körülmények között a regulációs rendszer révén szorosan kapcsoltak szétkapcsolódnak. Normál körülmények között a sejtek csak külsô jel, növekedési faktor hatására osztódnak, a tumorsejtek viszont bizonyos onkogének aktiválódása nyomán utánozni tudják ezeket a külsõ jeleket.
Egy egészséges felnôtt szervezetben normál
körülmények között egy-egy szövetben a
sejtek száma nagyjából állandó. A sejtszámot
és a specializált funkció fönntartását
kölcsönhatások sorozatán keresztül érvényesülô
endokrin, parakrin és mikrokörnyezeti reguláció
biztosítja. Az aktivált onkogénekkel rendelkezô
transzformált sejteken ezek a hatások felerõsödnek,
mivel a szervezet megpróbálja kontroll alatt tartani az "elszabadult"
sejteket. Ezért a transzformálódott sejtekre számos
hormon, növekedési és differenciálódási
faktorok, cytokinek, inhibitorok, valamint egyebek közt extracelluláris
mátrix proteinek, immunogén modulátorok stb. fejtik
ki hatásukat. A tumor kifejlôdése akkor kezdôdik,
amikor a transzformálódott sejtek tartósan osztódni
képesek ebben a számukra "ellenséges" mikrokörnyezetben,
és újabb genomiális változások begyûjtése
révén egyfajta, a szervezet szempontjából negatív
evolúciós folyamat eredményeként jönnek
létre az egyre életképesebb sejtvariánsok,
melyek többek közt pl. képesek elkerülni az immunrendszer
felismerô képességét, megszüntetik intercelluláris
kommunikációs csatornáikat és elkezdenek invazivitást
biztosító, illetve angiogenikus faktorokat termelni.
A programozott sejthalál és a halhatatlanság halála
A normál sejtek az intercelluláris kommunikációs kapcsolatok sérülése esetén, pl. az integrin, illetve egyéb extracelluláris mátrix proteinek jeleinek hiányában, apoptózis révén elpusztulnak, azaz megfogalmazható az a filozofikusan sem jelentéktelen (és talán az emberi társadalomra is általánosítható) tétel, mely szerint az élet alapmotívuma a kapcsolat, a kommunikáció, a szélsôséges individualizmus; a mimikált kapcsolat, ill. kommunikáció végül is pusztulásához vezet. Az intercelluláris kapcsolatok hiányában a sejtek többnyire a saját maguk által termelt "halál-molekulák" (death ligands) közvetítésével az apoptotikus kaszkád aktivációja révén pusztulnak el, ugyanakkor a genotoxikus, illetve egyéb súlyos sejtkárosító hatásoknak kitett sejtek ugyancsak programozott sejthalállal pusztulnak el. Ezt a hatást viszont fôleg a tumor szupresszor gének által kódolt regulátor fehérjék (p53, Rb stb.) közvetítik. A sejteknek ez az öngyilkos programja védi a sejtet és a szervezetet a mutációk begyûjtésétôl, a negatív evolúciós hatásoktól. A tumoros transzformáció során tehát az onkogének aktiválódása révén a sejtek folyamatosan fals proliferációs jeleket kapnak, másrészt a tumor szupresszor gének mutációi miatt nem mûködnek a repair, a sejtosztódás-gátlás, illetve a programozott sejthalál mechanizmusai. A tumorsejtek halhatatlanságát a tumor szupresszor gének dezaktiválása mellett számos egyéb negatív evolúciós folyamat is biztosítja, pl. az antiapoptotikus gének, illetve proteinek (bcl2, bclx stb.) aktivációja és a proapoptotikus gének, illetve proteinek (bad, bax) gátlása, a "sorsfonalat" folyamatosan újraépítô telomeráz enzim aktiválása és az apoptotikus gépezet végrehajtó enzimjeinek sérülése, illetve gátlása. A tumorsejtek szelektív elpusztítása tehát a halhatatlanságot biztosító antiapoptotikus mechanizmusokkal szembeni alternatív apoptotikus mechanizmusok beindítását igényli.
Az onkogének és tumor szupresszor gének "kooperativitása"
és a jeltovábbítási mechanízmusok zavarának
koncepciója alapvetôen új irányt adott a rák
gyógyszeres kezelésének megközelítésében.
Az új kutatási koncepció az onkogének és
a növekedési faktorok által létrehozott "fals",
illetve felerôsödött jelek, a mimikált kommunikáció
gátlását célozta meg. Ugyanakkor ma már
az is egyértelmû, hogy terápiás szempontból
nem elegendô a sejtosztódás gátlását
elérni, a tumorsejteket el kell pusztítani. A legújabb
eredmények bebizonyították, hogy a tumorsejtek szelektíven
elpusztíthatók az apoptózis, illetve a programozott
sejthalál indukálása révén, ami szoros
összefüggésben van a tirozin kináz enzimek gátlásával,
illetve a foszforilációs-defoszforilációs kaszkád
bizonyos szinergikus kölcsönhatásaival.
Jeltovábbítás-gátlás és tumorterápia
A jelátvitel fô mechanizmusának a fehérjék foszforilációját tekintik. A sejtfelszíni receptorokon keresztül érkezô jelek különbôzô fehérje foszforilációs kaszkádok közvetítésével jutnak el a sejtmagba, így nem meglepô, hogy az onkogének legtöbbje protein kinázokat kódol. Ezen szabályozási mechanizmus fontosságának általános elismerését mutatja, hogy 1992-ben az élettani és orvosi Nobel-díjat E.G. Krebsnek és E. H. Fischernek ítélték az intermedier anyagcserében és a jelátvitelben szerepet játszó fehérjék reverzibilis foszforilációjának tanulmányozásáért.
A növekedés szabályozásának jelátvitele specifikus tirozin kináz enzimekkel indul, amelyek G proteineket, szerin/treonin kinázokat (pl. protein kináz C, MAP kináz) és egyéb jelátvivô mechanizmusokat serkentenek. Tirozin kináz aktivitást mutatnak polipeptid növekedési faktorok transzmembrán receptorai és az onkogének által kódolt transzformáló fehérjék jelentôs része. Ez a tény azt sugallja, hogy a tirozin kinázok néhány alapvetô, sejtnövekedést szabályozó jelet közvetítenek, ily módon meghatározva a növekedés szabályozásának fô jelátvitéli mechanizmusát. A foszfotirozin foszfatázok szintén kulcsszabályozói a tirozin kinázok közvetítette jelátviteli folyamatoknak. A szerin/treonin kinázok (például MAP kinázok), illetve protein kináz C szintén fontos szerepet játszanak a jelátvitelben és számos fiziológiai folyamatot szabályoznak, többek között a sejtnövekedést és -differenciációt, valamint a tumorképzôdést. Míg a MAP kinázok nyugalomban lévõ sejtekben a citoplazmában találhatók és mitogenikus jelre, differenciáció indukciójára és különféle stresszhatásokra transzlokálódnak a sejtmagban jelátviteli komponenseket és transzkripciós faktorokat foszforilálva, addig a protein kináz C a nyugvó állapotban lévô sejtekben fôleg a "cytosole"-ban van jelen, azonban aktiváló stimulusra a "cytosole"-ból membránba történô áthelyezôdését számos sejttípusnál kimutatták, és az enzimaktiválódás korai lépésének tekintik, amely a sejtekben specifikus biológiai hatásokhoz vezet. A mitogenikus és a differenciált funkciókat stimuláló jelek számos esetben ugyanazokon a foszforilációs kaszkádokon keresztül fejtik ki hatásukat, és a legújabb eredmények alapján megfogalmazható az a koncepció, mely szerint a mitogenikus jelek szelektivitását egy úgynevezett "network" jeltovábbítás biztosítja, ami azt jelenti, hogy bizonyos jelek kooperatív kölcsönhatása, illetve együttállása szükséges egy adott sejtválasz eléréséhez (1).
Az a feltételezés, hogy a karcinogenezis a komplex jelátviteli mechanizmusokban bekövetkezô torzulásnak tulajdonítható, számos új stratégiát vet fel az új rákellenes gyógyszerek kifejlesztése terén. Az új rákellenes gyógyszerkutatás fôleg olyan gyógyszerek kifejlesztésére irányul, melyek specifikus jelátviteli mechanizmusokra hatnak. Miután a folyamatos sejtosztódási jelet generáló onkogének és növekedési faktor receptorok jelentôs része tirozin kináz, ezért a figyelem elsõsorban ezek gátlására irányult. Az utóbbi években világszerte igen intenzív kutatások folytak a sejtmembránon áthatolni képes kismolekulájú, illetve "peptidomimetikus" tirozin kináz gátlók elôállítása céljából.
A modern gyógyszerkutatás alapvetô stratégiai
feladata, hogy egy kiválasztott célmolekula, például
receptor tirozin kináz esetében bebizonyítsa ennek
a célmolekulának meghatározó szerepét
bizonyos patomechanizmusok, például rosszindulatú
daganatok kialakulásában. Ennek az úgynevezett "validálási"
folyamatnak a keretében különbözô immunhisztokémiai
és molekuláris biológiai technikákkal elôször
is meghatározzák a tirozin kinázok expressziós
profilját normál és tumoros szövetben. Amennyiben
egyfajta asszociatív kapcsolat kimutatható bizonyos fajta
tumorok és receptor tirozin kinázok között, nagyszámú
klinikai minta analízisével kell bizonyítani, hogy
az adott RTK-ok overexpressziója, illetve mutációja
negatív klinikai prognózissal jár együtt. Ezt
követôen egy sor molekuláris biológiai technika
segítségével történhet annak kiválasztása
és igazolása, hogy melyik tirozin kináz játszik
döntõ szerepet egy adott típusú tumor kialakulásában,
illetve mely tirozin kinázok gátlása, esetleg kiiktatása
vezet az adott tumor gátlásához. Az ilyen típusú
célmolekula igazolási ("target validation") kísérletek
célja az adott receptor tirozin kinázhoz kapcsolódó
jeltovábbítási mechanizmus szelektív megszüntetése
és így patológiás jelentõségének
bizonyítása.
Igen fontos in vivo validálási technikák még
a vizsgálandó gének szempontjából transzgenikus,
illetve ún. "knockout" egerek alkalmazása, és különbözô
humán örökletes betegségek tanulmányozása.
Az ily módon kiválasztott és igazolt célmolekulaként
szereplô tirozin kináz terápiás szempontból
releváns gátlására számos lehetõség
kínálkozik.
Tirozin kinázt gátló antitumor peptidhormonok
Az antitumor gyógyszer-hatóanyagok kutatásában az elôbbiekben vázolt szempontok alapján elôtérbe került új kutatási koncepció lényege, hogy bizonyos peptidhormonokból és ún. "peptidomimetikus" vegyületekbõl a szerkezet-hatás összefüggések tanulmányozása alapján kifejleszthetôk olyan szelektív, rákellenes vegyületek, amelyek beavatkoznak a tumorsejtek jelátadási folyamataiba és programozott sejthalált indukálnak. Számos adat bizonyítja, hogy az antitumor hatású peptid-hormon származékok közvetlenül specifikus receptorokon keresztül hatnak a rákos sejtekre, és így változtatják meg vagy gátolják a növekedési faktorok és a kapcsolódó onkogének jelátviteli mechanizmusait. Az általunk kifejlesztett TT-232 jelû szomatosztatin analóg jelentôs in vitro és in vivo antitumor aktivitással rendelkezik, jeltovábbítási mechanizmuson keresztül közvetve gátolja a tirozin kinázokat és programozott sejthalált indukál.
A szomatosztatinnak mint növekedést gátló faktornak az egyik legjelentôsebb hatása az, hogy a kóros sejtszaporodás különbözõ formáit képes befolyásolni. A szomatosztatin ugyanakkor egy általános antiszekréciós hormon, mely többek közt gátolja a glukagon, az inzulin és a növekedési hormon felszabadulását. A szomatosztatin mint endogén sejtosztódást gátló faktor és antiszekréciós hormon világszerte az érdeklõdés középpontjában áll. Különbözô kutatócsoportok tôbb száz analógot szintetizáltak a terápiás hatékonyság növelése, a biológiai stabilitás és a szelektivitás növelése céljából. Ma már egy szomatosztatin analóg klinikai gyakorlatban (Sandostatin), kettô pedig klinikai fejlesztés stádiumában van, de szelektív hatású szomatosztatin analógok elõállítása hosszú ideig nem igazán sikerült.
Mi azért kezdtünk el szomatosztatin analógok kutatásával foglalkozni, mert ismeretessé vált, hogy szomatosztatin analógok bizonyos tumorsejtekben aktiválják a tirozin foszfatázokat. A szerkezet-hatás összefüggések szisztematikus tanulmányozásával számos új analógot állítottunk elô, azzal a céllal, hogy növeljük a szelektív antiproliferatív hatást, ezért az analógokat elsôdlegesen tirozin kináz gátlásra és antiproliferatív hatásra teszteltük. Sikerült kifejleszteni és szabadalommal védeni egy olyan új, szelektiv biológiai hatású szomatosztatin analógot, mely erôs anfitumor hatást mutat számos humán tumor sejtvonalon, ugyanakkor a növekedési hormon [GH) felszabadulását nem befolyásolja. Ezt az analógot TT-232 név alatt vezettük be az irodalomba (2,3).
TT-232 szomatosztatin analógunk laboratóriumunkban vizsgálva erôs antitumor hatást mutatott számos tumor sejtvonalon (MCF-7 humán emlô és PC3 humán prosztata, ill. SW 620 és HT 29 humán vastagbél tumor sejteken), ugyanakkor a GH felszabadulást nem befolyásolta és nincs antiszekréciós hatása. Magmágneses rezonancia spektroszkópiával (NMR) kimutattuk, hogy a molekula különleges konformációs sajátságokkal rendelkezik a hagyományos szomatosztatin analógokhoz képest, és nem kötôdik a hipotalamuszban, a hipofízisben, illetve a cortexben, ugyanakkor jelentôsen kötôdik a tumorsejtekhez, ami alátámasztja korábbi, szelektivitásra vonatkozó vizsgálatainkat.
Kimutattuk, hogy a TT-232 igen széleskörû és dózisfüggôen dramatikus antiproliferatív hatása az apoptózis indukálásával függ össze, és az apoptotikus hatás közvetítésében a tirozin kinázok gátlásának fontos szerepe van, mivel a TT-232 hosszú távon gátolja a tirozin kinázokat, rövid távon viszont a foszfotirozin foszfatázok aktivitását stimulálja.
A TT-232 szomatosztatin analóg antitumor hatását
a National Cancer Inst. (USA) 60 humán tumor sejtvonalon tesztelte,
és 59-en hatásosnak bízonyult. In vivo antitumor vizsgálatokban
azt találtuk, hogy a TT-232 jelentôsen gátolja különbözô
állati tumor modelleken a tumor növekedését,
és igen jelentõs antimetasztatikus hatása van.
Peptidomimetikus tirozin kináz gátló molekulák
Az utóbbi években világszerte igen intenzív kutatások folytak a sejtmembránon áthatolni képes kismolekulájú "peptidomimetikus" (peptideket modellezô) tirozin kináz gátlók elôállitására. Ezen kutatások célja az onkogének által indukált jeltovábbítási kaszkád specifikus enzimjeinek, elsôsorban a receptor tirozin kinázoknak a szelektív gátlása.
Az elmúlt évek során szelektív tirozin kináz gátló molekulák után kutatva felépítettünk egy szisztematikusan tervezett molekula-könyvtárat, és ezeket a molekulákat különbözô receptor tirozin kinázok gátlására és programozott sejthalál indukálásra teszteltük. A tirozin kináz gátló molekulák tervezésének korábbi stratégiájával szemben nem az egyes enzimek szubsztrátkötõ helyeit vettük célba, hanem szelektív, ATP (adenozin-trifoszfá,. a sejt energiaháztartásában és jelátvitelében kulcsszerepet játszó molekula)-kötô helyen ható anyagokat kerestünk. A szerkezet-hatás összefüggések tanulmányozása alapján olyan "peptidomimetikus" tirozin kináz gátló vegyületeket állítottunk elô, amelyek különbözô ATP analóg farmakofor struktúrákat jártak körül. Molekuláinkat specifikus receptor tirozin kináz esszékben teszteltük, és a leghatékonyabb tirozin kináz gátlók hatását megvizsgáltuk in vitro és in vivo tumor növekedésre és az apoptózis jelenségére.
A közel 3000 molekulából álló molekula-könyvtárunk alapján, a fiziko-kémiai és biológiai adatok fölhasználásával QSAR (szerkezet-hatás összefüggés) és vezetô-molekula optimalizációs számításokat végeztünk, melyek alapján újabb molekulákat terveztünk és tervezünk. Sikerült több olyan molekulacsaládot is kifejlesztenünk, amelyek szelektíven gátolnak bizonyos receptor tirozin kinázokat, és jelentôs in vitro, illetve in vivo antitumor aktivitással rendelkeznek bizonyos tumor modelleken (4,5,6).
Az általunk elôállított szelektív EGF, illetve PDGF-receptor tirozin kináz gátló molekulák ICSO értékei 110 mM között voltak és két EGFR tirozin kináz gátló molekulánk IC50 értéke a szubmikromolos tartományban volt. Kontrollként elôállítottunk az irodalomban közölt legjobb EGF és PDGFR tirozin kináz gátló molekulák közül is néhányat, melyek IC50 értékei ugyanebbe a tartományba estek. TK gátló molekuláink jelentôs apoptózist indukáló hatással is rendelkeztek, és ezen molekulák esetében az apoptózist indukáló hatás jól korrelált a tirozin kináz gátló hatással. Mivel a TT-232 jelû szomatosztatin analógunk tirozin kináz gátló hatása és apoptózist indukáló hatása között szoros korreláció volt, megvizsgáltuk a leghatékonyabb kismolekulájú tirozin kináz gátló molekulák apoptózisra kifejtett hatását. Ezen vizsgálataink során azt találtuk. hogy az egyik EGF-receptoron ható, és kontrollmolekulának elôállított tirozin kináz gátló molekulánk egy, mindezidáig csak embrionális sejtekben kimutatott és nem indukálhatónak vélt programozott sejthalált okozott a tumorsejtekben, ami a tumorsejtek szelektív elpusztításának lehetôségét ígéri.
A szisztematikusan felépített molekula-könyvtárunkból
sikerült kiválasztanunk egy olyan flk1 tirozin kinázt
gátló, és programozott sejthalált indukáló
molekulát, ami in vitro és in vivo körülmények
között is jelentôs angiogenezis gátló hatású,
és mint ígéretes antitumor ágens preklinikai
fejlesztés alatt áll (5,6).
Flk1 tirozin kinázt és angiogenezist gátló antitumor ágensek
Az antitumor hatóanyagok kutatásának egyik legszéleseb spektrumú és legígéretesebb területe az angiogenezis gátlás, mivel folyamatos vérellátásra és ehhez új vérerek képzôdésére szinte valamennyi szilárd tumornak szüksége van. A tumorangiogenezis gátlására számos stratégiai lehetôség kínálkozik, amint azt a 2. ábra mutatja.
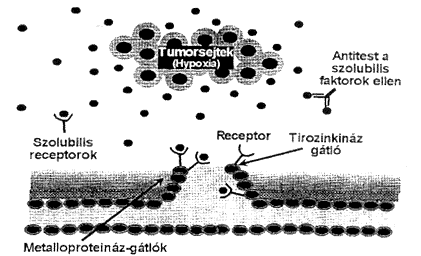
2. ábra. Angiogenezisgátló stratégiák
Az angiogenezis többlépéses folyamat, amelynek az eredménye új véredények formálása már meglévõ erekbõl. Angiogenezis során a faktorok által stimulált endothel sejtek olyan enzimeket termelnek, amelyek elbontják a bazális membránt, és így az endothel sejtek áthatolnak a membránon és folyamatos osztódásuk eredményeként új kapilláris ereket formálnak. Ez a folyamat elengedhetetlen a tumorok egy minimális méreten túli növekedéséhez. Számos angiogenikus növekedési faktor (jelenlegi ismereteink szerint 19) specifikus receptorokon keresztül fejt ki stimuláló hatást ennek az összetett folyamatnak különbözô lépéseire.
A fibroblast növekedési faktor (FGF) és az EGF receptorok a kemotaxishoz és az endoteliális sejtek mitogeneziséhez továbbítanak jeleket. A Vaszkuláris Endotheliális Növekedési Faktor (VEGF) receptorairól, az Flk1/KDR-rôl és az Flt1-rôl, in situ hibridizációval kimutatták, hogy kizárólag endothel sejteken expresszálódnak. Az Flk1 részt vesz a VEGF által indukált mitogenezisben, de az Flt1 szerepe még nem tisztázott. A tirozin kináz receptorok egy olyan új családját is megtalálták az endoteliális sejteken, amelyek Tie-et és Tek/Tie2-t tartalmaznak. Bár ezeknek a ligandumjait és pontos szerepét még nem azonosították, de az expresszió sémája és az egereken végrehajtott irányított mutagenezis eredményei azt sugallják, hogy részt vesznek az angiogenezis folyamatában.
Az endotéliumba történô restrikciós expressziója az Flk1-nek és humán analógjának, a KDR-nek is azt bizonyítja, hogy az Flk1 fontos szerepet játszik az angiogenezisben. In vivo domináns-negatív kísérletekben is kimutatták, hogy az Flk1 specifikus gátlása elnyomta rágcsálókban különbözô szolid tumoroknak az s.c, ill. intracerebrális növekedését. Úgy tûnt, hogy a mechanizmus az angiogenezis gátlása volt, azt jelezve, hogy szolid tumorokban az angiogenezisnek a VEGF/Flk-1 a fô útvonala. Ezt más eredmények is alátámasztották, amelyek azt mutatták, hogy tumorsejtek növekedését gátolni lehetett egerekben anti-VEGF monoklonális antitestekkel vagy anti-VEGF antiszenz RNS expresszióval.
Mindezek alapján egyértelmûvé vált, hogy Flk1/KDR inhibitorok kifejlesztése nagyon ígéretes területe az angiogenezishez kapcsolt betegségek és például a tumorok gyógyításának. Mi amerikai kutatókkal együttmûködve kezdtünk el foglalkozni olyan kismolekulájú tirozin kináz gátlók elôállításával, amelyek specifikusan gátolják az Flk1 kináz enzim aktivitását és a "downstream" eseményeket. Sikerült kifejlesztenünk több olyan molekulát is, amely hatékonyan és szelektíven gátolta az Flk1 tirozinkinázt, 1mM körüli IC50 értékkel. Az összes vegyület, amely gátolta az Flk1 kinázt, gátolta az endoteliális sejtek DNA-jába történô timidin inkorporációt is összemérhetô IC50 értékekkel. Ez alátámasztja azt az állítást, hogy az Flk1/KDR gátlása meggátolja az endoteliális sejtek más funkcióit is.
Összefoglalva megállapíthatjuk, hogy a modern tumorterápia kulcsfogalmai az intercelluláris kommunikáció, a fals jeltovábbítási rendszerek gátlása és a programozott sejthalál mechanizmusainak helyreállítása. Annak alátámasztására, hogy ez a három fogalom egymással is szorosan összefügg egy régi kínai bölcsességet szeretnék idézni:
Az élet s a halál egymást öleli át
A fény és a sötét kavargó táncra kél
Bennünk s köröttünk a lét
Együtt s egymásért él.
IRODALOM
1. Egan. S.E. and R.A. Weinberg (1993). The pathway to signal achievement.
Nature 365: 781783
2. Kéri Gy., l. Mezõ, A. Horváth, Zs. Vadász,
A. Balogh, M. Idei, T. Vántus, I. Teplán, M. Mák,
J. Horváth, K. Pál, O. Csuka, Novel Somatostatin analogs
with tyrosine kinase inhibitory and antitumor activity, Biochem. Biophys.
Res. Comm. 191: 681687. 1993
3. Kéri Gy., J. Érchegyi, A. Honváth, I. Mezõ,
M. Idei, T. Vántus, Zs. Vadász, Gy. Bökönyi, I.
Teplán, O. Csuka, M.Tejeda, D. Gaál, Zs. Szegedi, B. Szende,
C. Roze, H. Kalthoff and A. Ullrich, A Tumor-selective Somatostatin Analog
(TT232) with strong in vitro and in vivo antitumor activity. Proc. Natl.
Acad. Sci. (USA) 93:1251312518, 1996
4. Õrfi L., J. Kökösi, Gy. Szász, I. Kövesdi,
M. Mák, I. Teplán, Gy. Kéri, Heterocondensed Quinazolones:
Synthesis and protein-tyrosine kinase inhibitory activity. Bioorganic &
Medicinal Chemistry 4: 547551. 1996
5. Straun, L.M., G. McMahon, H. App, C. Tang, A. Levitzki, A. Gazit,
L. Chen. Gy. Kéri, L. Ôrfi, I. Flamme, A Ullrich, K.P.
Hirth and L.K Shauver, Flk1 as a target for tumor growth inhibtion. Cancer
Research 56: 35403545, 1996
6 Hirth. K.P., Schwartz D.P., Mann, E., Shauver, L. K., Kéri.
Gy., Székely. l., Bajor, T., Haimichael, J., Ôrfi, L., Leuitzki,
A., Gazit. A., Ullrich, A. and Lomers, R. Treatment of platelet derived
growth factor related disorders such as cancers. U.S. Patent No.: 5700822
(1997)