Karbokation-kutatásaim, a karbokationok
szerepe a kémiában
Oláh György Nobel-elôadása
1994. december 8-án
Sokan kutatták a hosszú élettartamú karbokationokat; a vizsgált problémakör gyorsan nôtt, és sikeresen lehetett tanulmányozni gyakorlatilag bármelyik karbokationrendszert. Nincs arra idô, hogy felsoroljam minden munkatársamat, sem azokat, akik a világ minden táján olyan sokat tettek ennek a területnek a fejlôdése érdekében. Munkásságuk megtalálható a kiterjedt irodalomban. Különösen ki kell emelnem azonban D.M. Brouwernek, H. Hogeveennek és munkatársainak az amszterdami Shell Laboratóriumban a hatvanas és hetvenes években végzett úttörô munkásságát. Alapvetô kutatásokat végeztek a hosszú élettartamú karbokationok tanulmányozása és az ehhez kapcsolódó szupersavas szénhidrogének kémiája területén. Az alkilkationokra vonatkozó, Shell Laboratóriumból származó közlemény 1964-ben jelent meg a Chemical Communications címû folyóiratban, nem sokkal az én 196264-es elsô beszámolóim után. Hasonlóan kell méltatni R.J. Gillespie alapvetô hozzájárulását az erôs savak (szupersavak) kémiájához, valamint azt az önzetlen segítséget, mellyel támogatott Kanadában a Dow-laboratóriumban. Ekkor kerültem vele újból kapcsolatba, mivel elôzôleg már találkoztunk 1956-ban Londonban a University College-ben, ahol ô C.K. Ingold munkatársa volt. Ezután költözött át Hamiltonba (Kanada, Ontario) a McMaster Egyetemre. Nála már az ötvenes évek végén mûködött NMR-spektrométer, és hozzásegített, hogy a karbokationok rendkívül erôsen savas rendszerét tartalmazó SbF5-öt vizsgálva az ô készülékén vehessük fel a spektrumokat. Az ô fluor-kénsav iránti érdeklôdése és a mi különbözô rendszereket tartalmazó SbF5-vizsgálataink volt a közös alap ahhoz, hogy tanulmányozzuk az FSO3H-SbF5 rendszereket. Azt is Gillespie javasolta, hogy a 100%-os kénsavnál erôsebb protikus savakat szupersavaknak nevezzük el. Ma már általánosan használják ezt az önkényes, de igen hasznos definíciót. Hangsúlyozni kell azonban, hogy maga a "szupersav" elnevezés a Harvardról, JB. Conanttól származik, aki 1927-ben így nevezte azokat a savakat, amelyek mint a perklórsav erôsebbek a hagyományos szervetlen savaknál, és még olyan gyenge bázisokat is képesek protonálni, mint a karbonilvegyületek. 1985-ben Surya Prakashsal és Jean Sommerrel írt "Szupersavak" címû könyvünket Conant emlékének ajánlottuk, habár ma már kevés kémikus ismeri az ezen a területen kifejtett munkásságát.
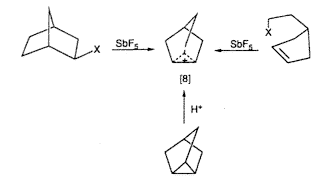
Ma is világosan vissza tudom idézni a már említett 1962. évi Brookhaven Mechanizmus Konferenciát, ahol elôször számoltam be nyilvánosan a hosszú élettartamú karbokationokról. A program szerinti "fô esemény" Saul Winsten és Herbert C. Brown (a bór-hidrogén-kémia úttörôje, Nobel-díj 1979) vitája volt a karbokationoknak (vagy ahogy akkor nevezték a karbóniumionoknak) klasszikus vagy nem klasszikus jellegérôl. Nagy meglepetés lehetett számukra, de az egész hallgatóság számára is, hogy egy ismeretlen ipari laboratóriumból jött fiatal vegyészt hívtak meg a fôelôadás megtartására, aki azt állította, hogy elôállított és vizsgált stabil, hosszú élettartamú karbóniumionokat (azaz karbokationokat), mégpedig igen egyszerû új módszerrel: nagyon erôs savas rendszerek (szupersavak) felhasználásával. Emlékszem, hogy Winstein és Brown külön-külön félrehívott, és figyelmeztetett, hogy egy fiatal vegyésznek rendkívüli módon meg kell gondolnia, mielôtt ilyen kijelentéseket tesz. Mindkettô azt hangsúlyozta, hogy valószínûleg tévedek, mert nem állíthattam elô hosszú élettartamú karbóniumionokat. Ellenben, ha a módszerem jó, akkor számomra lehetôség nyílik bebizonyítani, hogy a sokat vitatott 2-norbornil-kation "klasszikus" vagy "nem klasszikus". A nagy port felvert szembenállásuk középpontjában az a kérdés állt, vajon a 2-exo-, 2-endo-norbornil-észterek hidrolízisének kísérletileg tapasztalt jelentôs felgyorsulásának és a rendszer magas exo-szelektivitásának az oka mint azt Winstein állította a C1-C6 egyes kötés s-része, ahol a hidat képezô "nem klasszikus" ion delokalizációja is jelen van, vagy pedig csak az endo-rendszer és a hozzá tartozó kiegyensúlyozó "klasszikus" három vegyértékû ionok sztérikus gátlásáról van szó. A nem klasszikus ionok (a kifejezést elôször J.D. Roberts használta) P.D. Bartlett szerint nem tartalmaznak elég elektront ahhoz, hogy minden "kötésre" jusson egy pár belôlük, azaz arra van szükség, hogy legyen alapállapotban delokalizált s-elektronjuk. Mivel a módszeremmel a karbokationokat elô lehetett állítani, mint hosszú élettartamú képzôdményeket, megvolt az alkalom arra, hogy az ion közvetlen megfigyelésével kísérletileg döntsük el a kérdést. Ebben az idôben az SbF5-ben oldott 2-norbornil-fluoridnak még csak a szobahômérsékleti protonspektruma állt rendelkezésemre. Ezen viszont egy széles csúcs látszott, amely a hidrideltolódás és a WagnerMeerwein-átrendezôdés teljes egyensúlyára utalt (ez jól ismert a szolvolízises reakciókból és a 2-norbornil-rendszerek ezzel kapcsolatos átalakulásaiból). Kíváncsiságom nem hagyott nyugodni, ezért amikor 1964-ben átkerültem a Dow Eastern Laboratory-ba (amelyet Framinghamban létesítettek, Fred McLafferty igazgatósága alatt, és csak késôbb költözött Weylandba) a munkát együtt folytattuk Princetonból Paul Schleyerrel és a Yale-rôl Marty Saundersszel. Paul, akivel életre szóló barátságot kötöttem, már ekkor azzal a képességgel bírt, hogy mint valami katalizátor, képes volt közös erôfeszítéseket megszervezni. SO2-t használva oldószerül, le tudtunk menni az oldat hômérsékletével 78 oC-ig, és elôállítottuk az iont ciklopentenil-etil-fluorid ionizálásával vagy pedig nortricilén protonálásával FSO3H:SbF5/SO2ClF-ben [8].
Ekkor még mindig nem volt a megfelelõ NMR-mérésekhez szükséges, elég alacsony hômérsékletet biztosító berendezésem, de volt Marty Saundersnek. A mintáinkat ezért utaztattuk Massachusetts-bôl New Havenbe, ahol Saunders a Yale kémiai épületének alagsorában a maga építette NMR-készülékén egyre alacsonyabb hôfokokon vizsgálta a norbornilkation-oldatokat. Az ion spektrumát 70 oC-on is fel tudtuk venni, ahol már a 3,2-hidrid-eltolódás kifagyott. Csak akkor, amikor 1969-ben átköltöztem Clevelandbe a Case Western Reserve Egyetemre, fejleszthettem ki a megfelelõ alacsonyhômérséklet-technikát olyan oldószerek felhasználásával, mint az SO2CIF és SO2F2; így kaptam meg az egészen 159 oC-ig túlhûtött oldatokban a 2-norbornil-kation nagyfelbontású 1H- és 13C-NMR-spektrumát. Ilyen alacsony hömérsékleten be tudtuk fagyasztani mind az 1,2,6-hidrid-eltolódást, mind a WagnerMeerwein-féle átrendezôdést, és észlelhettük a statikus gyûrûs ionokat (3.a és 3.b ábra).
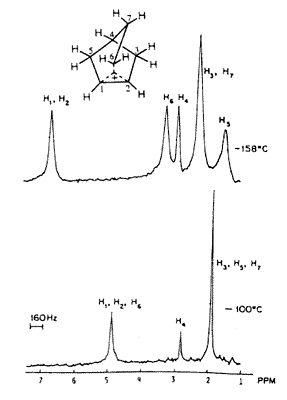
3.a ábra. 2-Norbornil-kation 395 MHz-es 1H-NMR-spektruma SbF5/SO2CIF/SO2F2-oldatban
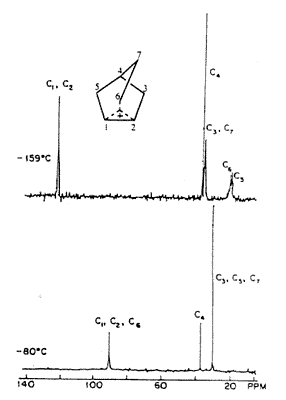
3.b ábra. (13C-vel dúsított) 2-norbornil-kation
50 MHz-es proton-lecsatolásos 13C-NMR-spektruma
SbF5/SO2CIF/SO2F2-oldatban
NMR-spektroszkópiával nehéz a gyûrûs nem klasszikus és a gyorsan egyensúlyba kerülô klasszikus karbokationokat egymástól megkülönböztetni, mivel az NMR viszonylag lassú, korlátozott idôskálájú fizikai módszer. Bizonyos részleteiben úgy oldottuk meg a problémát, hogy a két különbözô rendszer becsült eltolódásait használtuk, és összehasonlítottuk modellrendszerek adataival. Ma már a hatásos elméleti eljárások, mint az IGLO, GIAO nagyon megkönnyítik az egymástól különbözô ionok NMR-eltolódásainak kiszámítását és összehasonlítását a mérési adatokkal. Nagyon megnyugtató, hogy eredményeink és következtetéseink megállják a helyüket a legújabb, modern eszközökkel végzett vizsgálatok tükrében is.
Említettem, hogy már a karbokationos kutatásaink kezdetén (gyors vibrációs spektroszkópiai módszerrel) infravörös tanulmányokat is végeztünk. Felvettük a norbornilkation Raman-spektrumát is, és habár abban az idôben még nem lehetett a spektrumot elméletileg számítani a modell-vegyülettel (2-norbornilrendszer, illetve notriciklén) történt összehasonlítás alapján arra következtettünk, hogy az ion szimmetrikus, gyûrûs jellegû. Sunko és Schleyer újabb elegáns vizsgálataik során FT-IR-spektrumot vettek fel, és ezt hasonlították össze az elméleti számításokkal.
A másik eljárás, amellyel meg akartuk oldani a
gyûrûs, illetve a gyorsan egyensúlyba jutó ionok
kérdését Kai Siegbahn (fizikai Nobel-díj,
1981) elektronspektroszkópiája (ESCA) volt. A hatvanas évek
végén használtuk ezt karbokationok vizsgálatára
szupersavas mátrixban. Clevelandi laboratóriumomban George
Mateescu és Louise Riemenschneider helyezte üzembe
az ESCA-berendezést, ôk dolgozták ki az ESCA-spektrum
felvételének módszerét szupersavas mátrixban
több karbokationra, ideértve a terc-butil- és
2-norbornil-kationt (4. ábra). Ezek során újabb
meggyôzô bizonyítékokat kaptunk a 2-norbornil-kation
nem klasszikus jellegére, mivel az ESCA-spektrumban nem észleltünk
három vegyértékû karbénium-ioncentrumot,
azaz nem mutatott "klasszikus" ionjelleget, mint a terc-butil-kation
esetében. Az egyensúlyi klasszikus ionfogalom ellenzôi
részérôl ugyan újabb kritikák értek,
de Dave Clark vizsgálatai teljes mértékben
a mi eredményeinket és következtetéseinket igazolták.
Ezt bizonyította a kísérleti adatoknak az elméleti
számításokkal történô összehasonlítása
is.
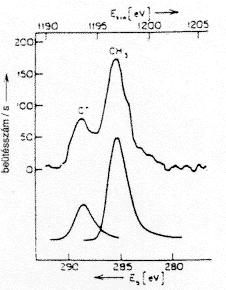 |
 |
| 4.a ábra. Terc-butil-kation szén 1s
fotoelektron-spektruma |
4.b ábra. 2-Norbornilkation 1s mag-
lyuk állapot spektruma és Clark szimu- lált spektruma klasszikus és nem klasszikus ionokra |
Illik megemlékezni néhány újabb jelentôs vizsgálatról is. Saunders eredményei azt bizonyították, hogy a lehetséges klasszikus egyensúlyi rendszerben nincs deutérium izotópeffektus. Nagyon alacsony (5 K!) hômérsékleten Myhre és Yannoni elôállítottak NMR-spektrumot szilárd állapotban, de azon semmi jelét nem találták az egyensúlyi klasszikus ionok kifagyásának. Ezen a hômérsékleten a korlátnak 200 cal/mol körül kell lennie (ez a vibrációs átmenet energiája). Laube viszont egykristályos röntgenszerkezet-vizsgálatot végzett szubsztituált 2-norbornil-kationokon. Schleyer az IGLO-t és hasonló számításokat is beleértve elméleti eszközökkel vizsgálta az NMR-eltolódást, adatainak összehasonlítása a kísérleti eredményekkel hozzájárult annak megértéséhez, hogy a 2-norbornil-kationnak 6-hidas karbóniumion jellege van. (A klasszikus 2-norbornil-kationt nem találták magasan fekvô közbensô terméknek!) Ugyanezt az eredményt adták Arnett kalorimetriás vizsgálatai is. Megállapítottuk az 1983-ban Prakashsal és Saundersszel írt, "A norbornilionnal kapcsolatos vita lezárása" (Conclusion of the Norbornyl Ion Controversy) címû cikkünkben, hogy "ezek a tanulmányok egyértelmûen véget vetettek az úgynevezett nemklasszikus-ion vitának". Winstein eredeti nézeteit egyértelmûen igazolták a széles körû, az én "stabilion-kémiámon" alapuló szerkezeti kutatások.
Sokan azt gondolják, hogy erre a problémára túl sok energiát pazaroltam, az én nézetem viszont az, hogy a norbornilion-vitának jelentôs hatása volt a kémiában. Nemcsak a szerkezetvizsgálatokhoz alkalmazott technikai módszerek határait segített kiterjeszteni, de lerakta a CH és CC egyes kötés, valamint a telített szénhidrogének elektrofil kémiája fejlôdésének alapjait is.
Az ellentétes vélemények alapos kritikai elemzése segít kiküszöbölni a lehetséges hibákat. Egyik kedvenc idézetem Békésy Györgytôl (Nobel-díj, 1961) származik: "A hibák kezelésének (egyik) módja az, ha van olyan barátunk, aki hajlandó idôt fordítani arra, hogy elôzetesen átnézze a kísérleteink terveit, majd az eredményeket, amikor a kísérleteket befejeztük. Még jobb azonban, ha van egy ellenségünk. Az ellenség képes rengeteg idôt és szellemi energiát áldozni arra, hogy kibogarássza kicsiny vagy nagy hibáinkat, és teszi mindezt ellenszolgáltatás nélkül. A baj az, hagy ritka az igazán tehetséges ellenség; a legtöbbjük csak olyan átlagos. Másik gond az ellenséggel, hogy néha barátunkká válik, és így sokat veszít a lelkesedésébôl. Jelen sorok szerzôje így veszítette el három legjobb ellenségét. Bárkinek, nemcsak tudósoknak, szüksége van egynéhány jó ellenségre!"
Az igaz, hogy nem volt hiány lelkes ellenfelekben (kicsit finomabb kifejezés ez, mint az ellenség), a norbornil-vita során mindkét oldalon. Nekik köszönhetô, hogy ma többet tudunk a karbokationok így a norbornilion szerkezetérôl, mint bármilyen más kémiai fogalomról. Az ô igyekezetük serkentett a vizsgálatok alaposabb elvégzésére, és sok kísérleti technika kifejlesztésére, illetve tökéletesítésére.
A norbornilkation-vizsgálatokból levont legjelentôsebb
következtetés számomra az, hogy a CH és CC
egyes kötés képes kételektronos s-donorként
szerepelni nemcsak intramolekuláris, de intermolekuláris
átalakulásokban és elektrofil reakciókban is.
Ezeknek a reakcióknak a kulcsa a kételektronosháromcentrumos
(2e3c) kötés (ez gyakori a bór- és fémorganikus
kémiában). Gyorsan kifejlôdött a kémia
sok új ága, és ez lehetôvé tette a hiperkoordinált
szénvegyületek (röviden hiperkarbon-vegyületek) kémiája
jelentôségének felismerését.
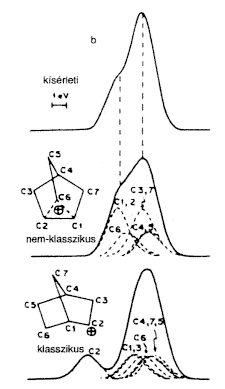
A karbokationok általános fogalma
A hosszú élettartamú megjelenési formák észlelése és a velük kapcsolatos szupersav-kémia alapozta meg a karbokationok kutatását, amelynek során világossá vált, hogy ezt a fogalmat tágabban kell értelmezni, mint korábban gondolták. Általánosabb definícióra van szükség; ezt én már egy 1972-ben megjelent közleményben felvetettem. A definíció azt veszi számításba, hogy a karbokationoknak két nagy, jól behatárolható osztálya létezik, és ezeket összeköti egy kontinuum, amely a delokalizáció különféle fokozatait tartalmazza.
a) A három vegyértékû ("klasszikus") karbéniumionokban sp2-hibridizációjú elektronhiányos szénatom van, amely általában planáris, ha ezt nem akadályozza a váz merevsége vagy sztérikus gátlás. (Meg kell azonban jegyezni, hogy az sp-hibridizációjú lineáris oxokarbónium-ionokban és vinilkationokban is elektronhiányos a szénatom.) A karbénium-szénnek hat vegyértékelektronja van, tehát nagymértékben elektronhiányos. A három vegyértékû karbokationok szerkezete mindig egyértelmûen leírható kételektronoskétcentrumos kötésekkel (Lewis-vegyérték kötésstruktúra). A három vegyértékû ionok alapképzôdménye a CH3+.
b) Ötös (vagy magasabb) koordinációjú ("nem klasszikus") karbónium-ionok, melyekben öt (vagy több) koordinált szénatom van. Ezeket nem írhatjuk le kizárólag kételektronoskétcentrumos egyes kötésekkel; itt szükséges a kételektronoshárom- (vagy több-) centrumos kötés bevezetése. A karbokation-centrumot mindig nyolc elektron veszi körül, de a karbóniumionok feltétlenül elektronhiányosak, mivel a két elektront három (vagy több) atommal osztják meg. A karbóniumionok alapképzôdményének a CH5+ tekinthetô.
Ezt követôen 1977-ben Brown és Schleyer javasolt újabb definíciót: "a nem klasszikus karbóniumion olyan pozitív töltésû kémiai egyed, amelyet nem lehet egyértelmûen egyetlen Lewis-struktúrával leírni. Az ilyen kation egy vagy több szén- vagy hidrogénhidat tartadmaz, amely két elektronhiányos centrumot köt össze. Az áthidaló atomok koordinációs száma nagyobb a szokásosnál, szénre általában öt vagy több, hidrogénre pedig kettô vagy annál nagyobb. Az ilyen ionokban kételektronos három- (vagy több ) centrumos kötések vannak, amelyeket szén- vagy hidrogénhíd kapcsol össze".
Lewisnek az a gondolata, hogy a kovalens kémiai kötést két atom között megosztott elektronpár képezi, a szerkezeti kémia sarokkövévé vált. A vegyészek hajlanak arra, hogy azokat a vegyületeket, amelyeknek a szerkezete nem írható le kizárólag ilyen kötésekkel, rendhagyóknak minôsítsék. Karbokationokban kevés az elektron ahhoz, hogy minden "kötéshez" egy párt adjanak, ezért ezeket "nem klasszikusként" határozták meg. Az elnevezést J.D. Roberts vezette be a ciklopropil-karbinol-kationra késôbb Winstein is alkalmazta a norbornilkationra. Még mindig használják ezt a nevet, habár már felismerték, hogy más vegyületekhez hasonlóan a szerkezetben kételektronos két- (vagy sok-) centrumos kötések szerepelnek (hasonlóan a Lipscomb (Nobel-díj, 1976) által a bórvegyületekre bevezetett kötési elvhez). Várható, hogy a "klasszikus" és "nem klasszikus" megkülönböztetés el fog tûnni, mivel fokozatosan rá fognak jönni, hogy itt a vegyértékkötés természetének általánosításáról van szó.
Hasznos a három vegyértékû karbéniumiont és pentakoordinált karbóniumiont mint szélsô egyedeket megkülönböztetni, azonban világosan kell látni, hogy a karbokationok rendszerében ezek között létezik a delokalizációnak több fokozata. Ezek tartalmazhatnak szomszédos n-donor-atomokat, p-donor-csoportokat vagy s-donor CH vagy CC kötéseket.
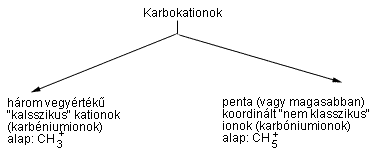
A három vegyértékû karbéniumionoknak mint intermediereknek kulcsszerepük van a telítetlen p-donor szénhidrogének elektrofil reakcióiban. A pentakoordinált karbóniumionok viszont a s-donor telített szénhidrogének (CH vagy CC egyes kötések) elektrofil reakcióihoz adják meg a kulcsot. Az egyes kötéseknek az a képessége, hogy elektrondonorként lépnek fel, onnan ered, hogy kételektronosháromcentrumos (2e3c) kötések kialakításával karbóniumionokat tudnak képezni.
A szénoktett kibôvítése 3d pálya segítségével nem látszik lehetségesnek, mivel a szénnek mint második periódusbeli kis rendszámú elemnek a külsô héján csak nyolc elektron lehet. A szén vegyértéke nem lehet négynél nagyobb. A Kekulé-féle négy vegyértékû szén fogalma kötéselméletileg azt jelenti, hogy négy atomhoz (vagy atomcsoporthoz) kapcsolódhat Lewis-féle 2e2c típusú kötéssel. Nincs azonban akadálya annak, hogy a szén részt vegyen többcentrumos kötésben, 2e3c (vagy multicentrumos) kapcsolódással. A szén penta- (vagy magasabb) koordinációjához öt (vagy több) atom vagy ligandum szimultán kapcsolódása szükséges, elfogadható kötéstávolságokon belül.
A szomszédos csoportok és egy karbéniumion-centrum hiányos p-pályája közösen stabilizálódnak olyan delokalizáció segítségével, amelybe bekapcsolódnak a kötésben részt nem vevô elektronpárral rendelkezõ atomok (n-donorok); a p-elektronrendszerek (direkt konjugáció vagy allil-stabilizálás), a hajlított s-kötések (mint a ciklopropil-karbinil-kationban) és a CH és CC s-kötéses hiperkonjugáció. A legegyszerûbb CH3+ kivételével a három vegyértékû karbéniumionokban ezért mindig megtalálható a delokalizáció többféle fokozata anélkül, hogy pentakoordinált karbóniumionná alakulnának. A határesetek képezik a karbokationok spektrumának a széleit.
A karbokationok szerepe az elektrofil reakciókban
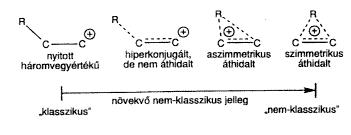
Savakkal katalizált elektrofil reakciókban és átalakulásokban: izomerizáció, alkilezés, szubsztitúció, addíció, elimináció, átrendezôdés stb. karbokationos közbensô termékek szerepelnek. Sok ilyen reakciónak jelentôs ipari felhasználása van. Az aromás szénhidrogénekre és az acetilénre alapozott kémia már egy évszázaddal ezelôtt megalapozta a szerves vegyipart. Késôbb az olefinekre épült vegyiparnak nôtt meg a jelentôsége. A kémia ezen ágaiban a reaktív p-kötéses rendszerek képezik az elektrondonor-szubsztrátumokat. Az elektrofil reakciókban ezek könnyen alkotnak három vegyértékû karbokationos közbensô termékeket (14. és 15. egyenlet).
Az imént említett pentakoordinált karbóniumionok felfedezése után felismerték azok fontos szerepét nemcsak a nem klasszikus ionok szerkezetének megértésében, de még lényegesebb ennél, hogy megadják a kulcsot a telített alifás szénhidrogénekben (alkánokban és cikloalkánokban) az egyes kötésen végbemenô elektrofil reakciókhoz. Ezekhez a reakciókhoz soroljuk nemcsak a savval katalizált szénhidrogén-izomerizálást, -fragmentációt, -ciklizációt, de a különféle szubsztitúciókat és a hasonló elektrofil reakciókat és átalakulásokat is.
A b-fenil-etil-rendszerek ionizációja során amely úgy tekinthetô, mint Cram-féle fenóniumionokat adó p-alkilezés a szomszédos p-járulék a karbokation-centrumhoz csatolódik. A 2-norbornil-rendszerek megfelelô ionizációja feltételez egy alkalmasan orientált CC egyes kötést (azaz intramolekuláris s-alkilezést), amely az áthidalt iont szolgáltatja.
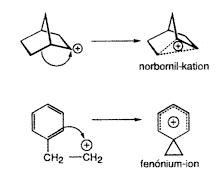
Továbbgondolva ezeket az összefüggéseket logikusan vetôdik fel a kérdés, miért nem befolyásolható a s-donor szénhidrogének intermolekuláris alkilezése (vagy más elektrofll reakció)?
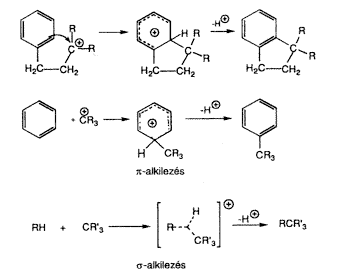
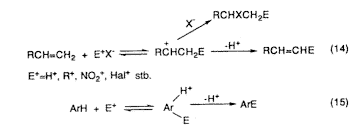
A hatvanas évek végén és a hetvenes évek elején a mi vizsgálataink szolgáltatták az elsõ bizonyítékokat arra, hogy a protolitikus folyamatokban, valamint a hidrogén-deutérium cserében, alkilezésben, nitrálásban, halogénezésben (16 és 17. egyenlet) az alkánok és cikloalkánok kovalens CH és CC egyes kötései általánosan reakcióképesek. A reakcióképesség annak köszönhetô, hogy az egyes kötések képesek s-donorként viselkedni (szigma-bázicitás), ami lehetôvé tészi, hogy a kötött elektronpárokat kételektronosháromcentrumos kötés képzése mellett megosszák más, elektronhiányos reagensekkel.
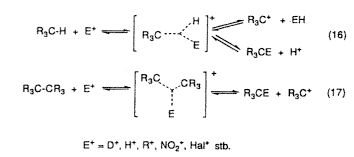
Az egyes kötés reakcióképessége ezek szerint onnan ered, hogy részt tud venni pentakoordinált karbóniumion kialakításában. A háromcentrumos kötés ezt követô felhasadása, a CH-reakció során eredményezi a szubsztitúciós termékeket; a CC-reakciókban hasad fel a kötés, karbéniumion-fragmens képzödik, ami azután további reakciókban vesz részt.
Az ötös koordinációjú karbóniumionokban
a kötéseltolódások könnyen, alacsony energiagáttal
mehetnek végbe, ezért a közbensô termékek
bonyolultabbak lehetnek, de mindig vannak köztük egymásba
alakítható karbóniumionok is.
Megnôtt a szupersavas szénhidrogén-kémia
ideértve a karbokation-intermedierek képzôdéseit
megkönynyítô feltételek gyakorlati felhasználásának
jelentôsége. Néhány említésre
méltó példa erre: alkánok viszonylag alacsony
hômérsékleten végrehajtott izomerizációja,
sokkal jobb és környezetkímélô alkilezés,
a metán felhasználásának új módszerei
és annak a lehetôsége, hogy felhasználjuk mint
magasabb szénhidrogének és származékaik
építôkövét hasonlóképpen
a szénnek kíméletesebb körülmények
között végrehajtott cseppfolyósítása.
Karbokation-rendszerek protonszolvatációs aktiválása
A karbokationok elektrofil, azaz elektronhiányos vegyületek. A telítetlen p-donor szénhidrogénekben és származékaikban (acetilén, olefinek, aromások) az elektrofil reagensekkel végbemenô reakciókat megkönnyíti a szubsztrátum nukleofil segítsége. Minél gyengébbek (dezaktiváltak) a reakciókban a p-donorok még inkább áll ez a csak gyenge elektronátadó képességgel rendelkezô telített szénhidrogénekre (s-donorokra) annál inkább magának az elektrofil anyagnak kell gondoskodnia a reakcióhoz szükséges hajtóerôrôl. Ezért szükségesek a nagymértékben elektrofil anyagok és a viszonylag kis nukleofilitással rendelkezô reakcióközegek (mint a szupersavas rendszerek).
Nem olyan nagyon régen értették meg, hogy az erôs
Brønsted- és Lewis-savakkal további kölcsönhatásra
(koordináció, szolvatáció) képes elektrofilek
erôsen aktiválhatók. A reakcióképességnek
ebbôl eredô megnövekedése nagyon jelentékeny
lehet, ha összehasonlítjuk a hagyományos körülmények
között reagáló egyszerû elektrofilekkel;
szuperelektrofil
alakzat jön létre, azaz olyan elektrofilek, amelyekben az elektronhiány
nagyon megnôtt (sokszor kétszeresen pozitív jelleggel).
Máshol
már beszámoltam a különbözô elektrofilek
szuperelektrofil aktivációjáról, ezért
most ezt nem szeretném tárgyalni, néhány olyan
szuperelektrofil aktiváció kivételével, amelyek
szintén hatással vannak a karbokationok reakcióképességére.
Az eredetileg Meerwein által tanulmányozott karboxóniumionokban egy alkilkationt így a terc-butil-kationt alkoxi-, például metoxicsoport helyettesít. A metoxicsoport (a szomszédos oxigén hozzájárulásával) delokalizálja a töltést, és így válnak ezek az ionok stabilabbakká.
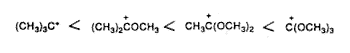
Csökkenthetô viszont a szomszédos oxigén hatása, ha az erôs sav-protoszolvát (vagy protonát) párosodik a nem kötött oxigénnel (18. egyenlet). Ebbôl következik, hogy szupersavas közegben a karboxóniumionok (és a rokon, például acilkationok) a szénen erôsen megnövekedett elektrofil reakcióképességet mutatnak, ami a dikationos jellegre utal.
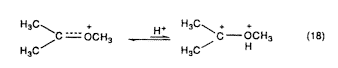
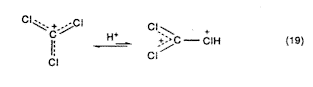
A halogén-szubsztituált karbokationok is (például a triklór-metil-kation, Cl3C+) erôsen stabilizálódnak (a BCl3-hoz hasonlóan) n-p-kölcsöhatással. Ezek protonáló szupersavas közegben jelentôsen aktiválhatók a halogén nemkötô elektronpárjával, amely csökkenti a szomszédos halogén hatását (19. egyenlet).
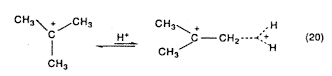
Az olyan kationokat, amelyekben csak a hiperkonjugált CH vagy CC egyes kötésektôl származó kölcsönhatások stabilizálják az elektronhiányos centrumot, a szupersavas szolvatáció aktiválja. Elméleti számítások, valamint deuterizált szupersavakban a hosszú élettartamú alkilkationok hidrogén-deutérium cseréje olyan körülmények között, amikor nem mehet végbe deprotonálásprotonálás, bizonyították, hogy ezek a protoalkilkationok a valódi intermedierek (20. egyenlet).
Az alkánoknak a jól ismert nagy reakcióképessége
az erôs savas közegben végbemenô izomerizációs
alkilezési reakciókban nagy valószínûséggel
az intermedier alkilkationok protonszolvatációjának
tulajdonítható. Hasonló aktiváció felléphet
olyan más, savval katalizált szénhidrogén-átalakulásokban,
amelyek fôleg olyan oldatokban játszódnak le, ahol
az oldatban savfelesleg van.
Aktiválás szilárd szupersavakkal, és ezek valószínû szerepe enzimes rendszerekben
A karbokationok kémiáját és aktiválásukat eddig csak szupersavas oldatokban tárgyaltuk. Szupersavas rendszerek azonban nem csupán oldatok kémiájával kapcsolatban léteznek. Egyre nagyobb a jelentôsége az olyan szupersavaknak, ahol mind Brønsted-, mind Lewis-savrégiók elôfordulnak. A skála a felvitt vagy közberétegezett rendszerektõl az erôsen savas, perfluoros rezinszulfonsavakig (mint a Nafion-H és analógjai) és a zeolitokig (például H-ZSM-5) terjed.
Ennek a figyelemre méltó aktivitásnak a magyarázatára amely például az alkánok (sôt a metán) katalitikus átalakulásában is fellép meg kell becsülnünk az ilyen szilárd savakban található savas régiók de facto aktivitását.
Ismeretes, hogy a Nafion-H-ban összecsomósodott (klaszteres) savas SO3H-csoportok vannak (5. ábra).
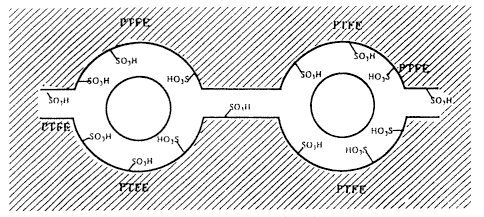
5. ábra. Klaszteres SO3H-sav helyek a perfluoriált rezinszulfonsavban
Haag és munkatársai azt találták, hogy a szintén szupersavas aktivitást mutató H-ZSM-5 könnyen izomerizálja és alkalizája az alkánokat (megfigyelték, hogy sztöchiometrikus mennyiségben H2 is fellép mint protolitikus melléktermék). Ebben a zeolitban szintén vannak egymáshoz nagyon közel körülbelül 25 nm távolságra aktív Brønsted- és Lewis-savrégiók (6. ábra).
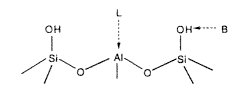
6. ábra. Brønsted- (B) és Lewis-sav- (L) helyek zeolitokban
Az oldatkémia analógiájára elfogadható, hogy ezekben (vagy más) szupersav-katalitikus rendszerekben kétszeresen vagy többszörösen egymásba illeszkedô ("egymásba fogazott") kölcsönhatások lehetségesek, amelyek folyamán nagyon reakcióképes közbensô termékek képzôdnek. Ez a protonszolvatáció (protonálás) szilárd fázisú megfelelôje.
A természet képes olyan átalakulások véghezvitelére,
amelyeket a vegyészek csak most kezdenek el megérteni, de
még nem tudnak reprodukálni. Enzimes tartományokban
sok olyan jelentôs átalakulás megy végbe, amelyeket
savak katalizálnak (idesoroljuk az elektronhiányos fémionokkal
katalizált folyamatokat is). Az enzimhelyek különleges
geometriája miatt lehetségesek egymásba fogazott kölcsönhatások;
a most tárgyalt fogalmak hozzásegíthetnek bizonyos
enzimes folyamatok megértéséhez.
Következtetések
A világ minden táján dolgozó kutatók hozzájárulásával nagyon aktív és gyors fejlõdést tapasztalhatunk a hosszú élettartamú karbokationok kémiája területén. Érthetô, hogy meg vagyok elégedve, ha az elért haladást és az elôttünk álló lehetôségeket veszem számba. Úgy indult ez, hogy a kémiai reakciók egyik legfontosabb közbensô termékeit (a karbokationokat) akartuk hosszú élettartamú alakban elôállítani, és szerkezetüket tanulmányozni. Ez azután elvezetett az egyes kötések (mint a CH és CC kötés) elektrofil reakcióképességének és az ezzel összefüggô szupersav-szénhidrogénkémia általános koncepciójához.
Annak ellenére, hogy igen nagy a haladás, mégis azt hiszem, hogy a legizgalmasabb szakasz még a kutatók jövô generációjára vár. Kívánom nekik, hogy olyan izgalmas és kielégítô legyen a munkájuk, mint amilyen az enyém volt.
Kekulé egy évszázaddal ezelôtt megfogalmazta a szén négyvegyértékûségének tételét és ez a szerves kémia alapját fogja ezután is képezni. A szén mint kis, második periódusbeli elem nem tudja kibôvíteni a külsô elektronhéját, az oktettszabály viszont nem enged meg csak olyan Lewis-típusú (vagy ezzel egyenértékû többszörös) kötéseket, ahol négy, kételektronoskétcentrumos (2e2c) kapcsolat alakul ki. Lehetséges azonban, hogy a szén egy (vagy több) elektronpárja kételektronosháromcentrumos (2e3c) kötésekbe kapcsolódik be. Ez lehetôvé teszi a szén egyidejû kötôdését öt (vagy akár hat) atomhoz vagy atomcsoporthoz. A "hiperkarbont" (hiperkoordinált szenet) tartalmazó karbóniumionnál éppen ez a helyzet. Ez adja a kulcsot a telített szénhidrogének beleértve az alapvegyület, metán és általában a CC és CH kötések elektrofil kémiája gyors fejlôdésének megértéséhez. Míg a hiperkoordinált karbokationok olyan 8e szénrendszerek, amelyek nem sértik az oktett-szabályt, addig a karbonionos [YCRX] SN2 átmeneti állapotok 10e-rendszerek, tehát nem lehetnek intermedierek.
Több, mint húsz évvel ezelôtt írtam
a következôt: "Egy napon annak a felismerésnek, hogy
a kötésben részt vevô (közös) elektronpárok
(egyes kötések) elektrondonorok, ugyanolyan fontosságot
fognak tulajdonítani, mint a Lewis-féle elképzelésnek
a magányos (kötésben részt nem vevô) elektronpárok
elektrondonor jellegérôl (idesorolnám még a
többszörös kötésben részt vevô
elektronpárokat is). Ma nemcsak magyarázni tudjuk a telített
szénhidrogének és egyes kötések reakcióképességét
az elektrofil reakciókban, de ezt az ismeretünket fel is tudjuk
használni arra, hogy a karbokation-kémiának új
területeit és új reakcióit fedezzük fel."
Bizonyos elégtételt érzek azért, hogy ez az
ígéret teljesült.
Köszönetnyilvánítás
Abban a szerencsében volt részem, hogy sokan mások
elkészítették azt az alapot, amelyre építhettem.
Elsôsorban Hans Meerweinrôl (18791965) és Christopher
Kelk Ingoldról (18931970) kell nagy háIával megesnlékeznem,
akik felismerték a karbokationok szerepét egyes kémiai
reakciókban, valamint Frank Whitmore-ról (18871947),
aki ennek általános jellegét állapította
meg. Nagyon hálás vagyok korábbi hallgatóimnak
és segítôtársaimnak, akiknek az odaadó,
kemény munkája, tudományos közremûködése
lehetôvé tette közös sikereinket.
Feleségem, Judy, nyugdíjba vonulásáig,
integrális részt vállalt tudományos erôfeszítéseinkben,
ugyanakkor az Oláh-csoportban mindenki nagyra becsülte melegszívûségéért,
azért a gondoskodásért, amellyel körülvette
a mi "tudományos családunkat". Felbecsülhetetlen
G.K. Prakash segítsége, aki kitûnô diplomája
megszerzése után kezdôdött együttmûködésünk
20 éve alatt nemcsak kollégám volt, de közeli
barátommá is vált. A Dél-kaliforniai Egyetem,
Loker Szénhidrogén-kutató Intézete otthont
és csodálatos támogatást nyújtott munkámhoz
az utóbbi 15 évben. Köszönöm az intézetbeli
munkatársaknak, fôképpen Mrs. Katherina Lokernek,
Harold
Moultonnak és Carl Franklinnek az évek során
nyújtott segítségüket és barátságukat.
Nagyra becsülöm az együttmûködést csodálatos
kollégáimmal Ned Arnettel, Joseph Casanovával,
Paul
Schleyerrel, Peter Stanggal,
Ken Wade-del és
Robert
Williamsszel, a Loker-intézet kitûnô idõsebb
kutatóival. Az Állami Egészségügyi Intézet
és a Tudományos Alap éveken keresztül adott támogatást
a karbokation-kutatásainkhoz; a Loker-intézettôl kaptuk
a legnagyobb segítséget szénhidrogén-kémiai
munkánkhoz.
| Vissza | http://www.kfki.hu/chemonet/
http://www.ch.bme.hu/chemonet/ |