""Ebben a módszerben megvan az a pontosság, ami az eddigiekbôl hiányzott, így nagyobb rendszerek vizsgálatára is alkalmas."
Néha a fehérjék olyanok, mint a Gonosz Boszorkány. Önts rá vizet, várj pár nanoszekundumot, és máris hallhatod a szánalmas nyögdécselést. "Olvadoook!" Hát, persze nem teljesen. De esetleg valóban hallható a biokémikusok nyögése, amint épp a vízzel érintkezô nagy fehérjéken vagy a DNS-en végzett szimulációjuk eredményét vizsgálják. Pár száz nanoszekumdumnyi szimulációs ideig minden jól megy, de azután a molekula szétesik. Ez nagyon bosszantó probléma, de úgy tûnik, hogy Tom Dardennek és Lee Pedersennek (Pittsburg Supercomputing Center) sikerült megoldást találnia rá.
A probléma molekuladinamikai (MD), azaz olyan számításokkal kapcsolatos, amelyek szimulálják és megjósolják, hogyan változik idôben a molekulák szerkezete. Ahogy az utóbbi 15 évben a számítástechnika egyre hatékonyabb lett, úgy vált az MD is a molekuláris biológiai eszköztár fontos részévé. A biológiakönyvek színes ábrái olyan fehérjéket ábrázolnak, amelyeket gondosan eltávolítottak a sejtbôl, majd kikristályosították ôket, hogy röntgen-krisztallográfiával meg lehessen határozni a szerkezetüket. Ezek az ábrák igen fontosak tudásunk gyarapításához, de csupán annyira reprezentálják a valóságot, mint a múzeumi tárlóban kiállított lepke. Az MD lehetôvé teszi – hogy az elôbbi hasonlatnál maradjunk –, hogy a lepke szálljon; a kimerevített molekulák csupán kezdôképek abban szimulációban, amelyben a kutatók a sejten belüli környezetben tudják vizsgálni a mozgásokat. A legvalószerûbb MD-szimulációk már a molekula körüli vízmolekulákat és ionokat is számításba veszik a vizsgálat során, így nyújtva minél valószerûbb képet a sejten belül ható erôkrôl.
Mindazonáltal minden egyes, a vizsgált molekulán belüli atom egymásra hatásának, valamint ezen atomoknak a vízzel való kölcsönhatásának a vizsgálata igen bonyolult számítási feladat. De ahogy a számítástechnikai eszköztár egyre hatékonyabbá vált, úgy vált egyre inkább elérhetôbbé és kívánatosabbá az, hogy nyomon követhessük, hogyan változnak ezek az alakzatok akár nanoszekundumokig is – ami kevesebb, mint egy szempillantás, de egy fehérje-biokémikusnak bôven elég. Egy ilyen szimuláció több száz számítógép-óráig is eltarthat, és az eredmény kiábrándító lehet.
Pedersen és Darden pár éve találkoztak elôször a fehérjeolvadással. "Az általunk vizsgált fehérje pár száz pikoszekundum alatt szó szerint darabokra rázta szét magát" – mesélte Darden, a National Institute of Enviromental Health Science biomatematikusa. "Ez az olvadás jóval erôteljesebb a DNS-nél. Úgy is mondhatjuk, minél tovább tart a szimuláció, annál rosszabb a helyzet."
A Pittsburgh Supercomputing Centerben végzett szimulációkkal sikerült behatárolni a baj okozóját: az elektrosztatikus erôket, azaz a vonzó-taszító kölcsönhatásokat a kötetlenül maradt atomok között. Darden még megoldást is talált. Egy új módszert dolgozott ki, az "Ewald-féle részecskehálót", amely elég gyors és ezért alkalmas nagyobb alakzatok vizsgálatára is. "Két fontos eredményünk van" – állítja Pedersen, a University of North Carolina fiziko-kémikusa. "Ebben a módszerben megvan az a pontosság, ami az eddigiekbôl hiányzott, így nagyobb rendszerek vizsgálatára is alkalmas." A módszer további elônyeinek felkutatására elkészítették a program párhuzamos változatát is, amely a CRAY T3D-n fut, Pittsburgh-ben.
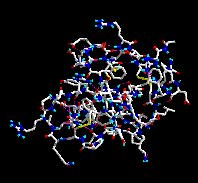
Az ábra egy nagy fehérje, a szarvasmarha hasnyálmirigyében termelt tripszin inhibitor molekuláris szerkezetét mutatja Darden és Pedersen Ewald-részecskehálós szimulációja alapján. A piros szín az oxigénmolekulákat jelöli, a kék a nitrogént, a világoskék a hidrogént, a fehér a szenet, a sárga a ként.
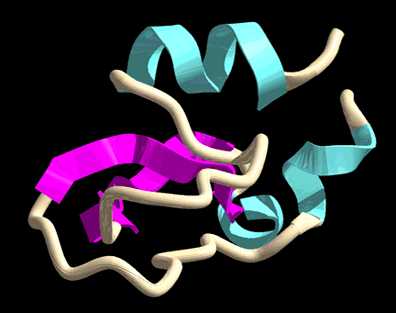
A tripszin gerince a másodlagos struktúra szerint: a véletlenszerû lánc (lisztfehér) csô alakú; a hélix (vízszínû) lapos, a burkolók (bíborvörös) pedig nyilak. A pöttyözött felület az oldószer számára hozzáférhetô felületet jelzi.
[ Vissza a válogatás listájára ]